
Abstract
The chemoselective bonding of a bifunctional organic molecule on a semiconductor surface is analyzed with density functional theory (DFT). Periodic energy decomposition analysis is used to reveal the bonding characteristics of different adsorption modes and transition states for 5-ethoxymethyl-5-methylcyclooctyne on Si(001). This system has previously been experimentally proven to be a prototype model system for inorganic–organic hybrid interfaces. The molecule thereby poses challenges for a theoretical description of conformational flexibility and competitive adsorption behavior of the two functional groups. We find that adsorption via the strained triple bond is preferred over the ether group, thus confirming previous experiments. Bonding analysis in combination with static DFT as well as ab initio molecular dynamics methods thereby reveals the determining factors for this chemoselectivity and shows that the functional groups barely influence each other in their surface adsorption.




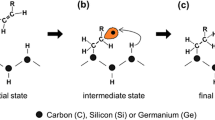
Part b reproduced with permission from Ref. [10]. Copyright John Wiley & Sons, Inc




Cyclooctyne data taken from Ref. [11]


Similar content being viewed by others

Notes
Minimum mode following algorithms, such as the Dimer method, are based on the premise that there is only one low-frequency vibration, i.e., the one that is going to invert its curvature. Climbing-image NEB, on the contrary, only works if the frequencies of the imaginary vibration and the lowest real vibration are large enough so that the computation is unsusceptible to numerical noise. Both methods failed to convert to first-order saddle points in this system.
References
Wolkow RA (1999) Annu Rev Phys Chem 50:413
Teplyakov AV, Bent SF (2013) J Vac Sci Technol A 31:050810
Rosenow P, Jakob P, Tonner R (2016) J Phys Chem Lett 7:1422
Hossain MZ, Yamashita Y, Mukai K, Yoshinobu J (2004) Chem Phys Lett 388:27
Reutzel M, Münster N, Lipponer MA, Länger C, Höfer U, Koert U, Dürr M (2016) J Phys Chem C 120:26284
Mette G, Dürr M, Bartholomäus R, Koert U, Höfer U (2013) Chem Phys Lett 556:70
Mette G, Reutzel M, Bartholomäus R, Laref S, Tonner R, Dürr M, Koert U, Höfer U (2014) ChemPhysChem 15:3725
Reutzel M, Mette G, Stromberger P, Koert U, Dürr M, Höfer U (2015) J Phys Chem C 119:6018
Reutzel M, Lipponer M, Dürr M, Höfer U (2015) J Phys Chem Lett 6:3971
Pecher J, Schober C, Tonner R (2017) Chem Eur J 23:5459
Pecher L, Schmidt S, Tonner R (2017) J Phys Chem C 121:26840
Pecher L, Laref S, Raupach M, Tonner R (2017) Angew Chem Int Ed 56:15150
Kresse G, Hafner J (1993) Phys Rev B 47:558
Kresse G, Hafner J (1994) Phys Rev B 49:14251
Kresse G, Furthmüller J (1996) Phys Rev B 54:11169
Kresse G, Furthmüller J (1996) Comput Mater Sci 6:15
Blöchl PE (1994) Phys Rev B 50:17953
Kresse G, Joubert D (1999) Phys Rev B 59:1758
Perdew JP, Burke K, Enzerhof M (1996) Phys Rev Lett 77:3865
Perdew JP, Burke K, Enzerhof M (1997) Phys Rev Lett 78:1396
Grimme S, Antony J, Ehrlich S, Krieg H (2010) J Chem Phys 132:154104
Grimme S, Ehrlich S, Goerigk L (2010) J Comput Chem 32:1456
Boyd DRJ (1955) J Chem Phys 23:922
Jónsson H, Mills G, Jacobsen KW (1998) In: Berne BJ, Ciccotti G, Coker DF (eds) Classical and quantum dynamics in condensed phase simulations. World Scientific, Singapore, p 385
Henkelman G, Uberuaga BP, Jónsson H (2000) J Chem Phys 113:9901
Henkelman G, Jónsson H (1999) J Chem Phys 111(15):7010
Raupach M, Tonner R (2015) J Chem Phys 142:194105
te Velde G, Baerends EJ (1991) Phys Rev B 44:7788
BAND (2016) SCM, theoretical chemistry, Vrije Universiteit, Amsterdam, The Netherlands. http://www.scm.com. Accessed 20 Feb 2018
Verlet L (1967) Phys Rev 159:98
Swope WC, Andersen HC, Berens PH, Wilson KR (1982) J Chem Phys 76:637
Nosé S (1984) Mol Phys 52:255
Nosé S (1984) J Chem Phys 81:511
Random decimal fraction generator. http://www.random.org/decimal-fractions. Accessed 20 Feb 2018
Pecher J, Tonner R (2017) ChemPhysChem 18:34
Pecher J, Mette G, Dürr M, Tonner R (2017) ChemPhysChem 18:357
Weigend F, Ahlrichs R (2005) Phys Chem Chem Phys 7:3297
Riplinger C, Neese F (2013) J Chem Phys 138:034106
Riplinger C, Sandhoefer B, Hansen A, Neese F (2013) J Chem Phys 139:134101
Kim S-W, Lee J-H, Kim H-J, Cho J-H (2013) Chem Phys Lett 557:159
Kuze N, Kuroki N, Takeuchi H, Egawa T, Konaka S (1993) J Mol Struct 301:81
Ess DH, Jones GO, Houk KN (2008) Org Lett 10:1633
Schoenebeck F, Ess DH, Jones GO, Houk KN (2009) J Am Chem Soc 131:8121
Chenoweth K, Chenoweth D, Goddard WA III (2009) Org Biomol Chem 7:5255
Pigge FC (2016) Curr Org Chem 20:1902
Acknowledgements
We thank HRZ Marburg, LOEWE-CSC Frankfurt and HLRS Stuttgart for providing computational resources and Jan-Niclas Luy (Marburg) for preliminary work.
Funding
We thank the Deutsche Forschungsgemeinschaft (DFG) for funding via SFB 1083
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Published as part of the special collection of articles “First European Symposium on Chemical Bonding”.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pecher, L., Tonner, R. Computational analysis of the competitive bonding and reactivity pattern of a bifunctional cyclooctyne on Si(001). Theor Chem Acc 137, 48 (2018). https://doi.org/10.1007/s00214-018-2212-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-018-2212-5