
Abstract.
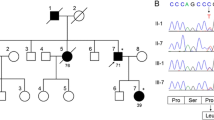
We describe three patients from two families with progressive spinocerebellar ataxia, peripheral neuropathy, raised α-fetoprotein (AFP) and hypercholesterolaemia. Two siblings had identical clinical features, with late childhood onset of symptoms and slow progression, requiring crutches to walk at ages 37 and 38 years. Another patient developed ataxia aged 13 years and became wheel-chair bound by 20 years of age. Although they all had raised serum AFP levels, their clinical, immunological, biochemical, cytogenetic and molecular genetic studies failed to support a diagnosis of Ataxia Telangiectasia. Extensive investigation including imaging, biochemical and genetic studies excluded other known ataxias. Their clinical features most closely resemble the phenotype of a single consanguineous Japanese family with four individuals affected by spinocerebellar ataxia, peripheral neuropathy, raised AFP and hypercholesterolaemia. Homozygosity mapping has identified a locus in this Japanese family at 9q34. Haplotype analysis of our cases demonstrated possible linkage to 9q34, suggesting these may be the first Caucasian families described with this disorder.
Similar content being viewed by others


References
Baraitser M (1997) The Genetics of Neurological Disorders. Oxford Monographs on Molecular Genetics-34. Third ed. Oxford University Press, Oxford, pp 146–165
Gatti RA (1998) Ataxia-Telangiectasia. In: Vogelstein BKK (ed) The Genetic Basis of Human Cancer. New York, Mc-Graw-Hill, pp 275–300
McConville CM, Stankovic T, Byrd PJ, McGuire GM, Yao QY, Lennox GG, Taylor MR (1996) Mutations associated with variant phenotypes in ataxiatelangiectasia. Am J Hum Genet 59:320–330
Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM (1999) The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 99:577–587
Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, Stockton DW, Lupski JR (2002) Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet 32:267–272
Aicardi J, Barbosa C, Andermann E, Andermann F, Morcos R, Ghanem Q, Fukuyama Y, Awaya Y, Moe P (1988) Ataxia-ocular motor apraxia: a syndrome mimicking ataxia-telangiectasia. Ann Neurol 24:497–502
Nemeth AH, Bochukova E, Dunne E, Huson SM, Elston J,Hannan MA, Jackson M, Chapman CJ, Taylor AM (2000) Autosomal recessive cerebellar ataxia with oculomotor apraxia (ataxiatelangiectasia-like syndrome) is linked to chromosome 9q34. Am J Hum Genet 67:1320–1326
Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, Koike R, Hiroi T, Yuasa T, Awaya Y, Sakai T, Takahashi T, Nagatomo H, Sekijima Y,Kawachi I, Takiyama Y, Nishizawa M, Fukuhara N, Saito K, Sugano S, Tsuji S (2001) Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet 29:184–188
Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T,Mendonca P, Costa M, Barros J, Yanagisawa T, Watanabe M, Ikeda Y, Aoki M, Nagata T, Coutinho P, Sequeiros J, Koenig M (2001) The gene mutated in ataxiaocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet 29:189–193
Watanabe M, Sugai Y, Concannon P, Koenig M, Schmitt M, Sato M, Shizuka M, Mizushima K, Ikeda Y, Tomidokoro Y, Okamoto K, Shoji M (1998) Familial spinocerebellar ataxia with cerebellar atrophy, peripheral neuropathy, and elevated level of serum creatine kinase, gamma-globulin, and alpha-fetoprotein. Ann Neurol 44:265–269
Bomont P, Watanabe M, Gershoni-Barush R, Shizuka M, Tanaka M, Sugano J, Guiraud-Chaumeil C, Koenig M (2000) Homozygosity mapping of spinocerebellar ataxia with cerebellar atrophy and peripheral neuropathy to 9q33–34, and with hearing impairment and optic atrophy to 6p21–23. Eur J Hum Genet 8:986–990
Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenolchloroform extraction. Anal Biochem 162:156–159
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215
Fensom AH, Benson PF, Crees MJ, Ellis M, Rodeck CH, Vaughan RW (1983) Prenatal exclusion of homocystinuria (cystathionine beta-synthase deficiency) by assay of phytohaemagglutinin-stimulated fetal lymphocytes. Prenat Diagn 3:127–130
Vanagaite L, James MR, Rotman G, Savitsky K, Bar-Shira A, Gilad S, Ziv Y, Uchenik V, Sartiel A, Collins FS, Sheffield VC, Richard III CW, Weissensbach J, Shiloh Y (1995) A highdensity microsatellite map of the ataxia-telangiectasia locus. Hum Genets 95:451–454
Izatt L, Vessey C, Hodgson SV, Solomon E (1999) Rapid and efficient ATM mutation detection by fluorescent chemical cleavage of mismatch: identification of four novel mutations. Eur J Hum Genet 7:310–320
Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S,Uziel T, Sfez S, et al. (1995) A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268:1749–1753
Taylor AM, Flude E, Laher B, Stacey M, McKay E, Watt J, Green SH, Harding AE (1987) Variant forms of ataxia telangiectasia. J Med Genet 24:669–677
Hassin-Baer S, Bar-Shira A, Gilad S, Galanty Y, Khosravi R, Lossos A, Giladi N, Weitz R, Ben-Zeev B, Goldhammer Y, Shiloh Y (1999) Absence of mutations in ATM, the gene responsible for ataxia telangiectasia in patients with cerebellar ataxia. J Neurol 246:716–719
Caldecott KW (2003) DNA singlestrand break repair and spinocerebellar ataxia (Review). Cell 112:7–10
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Izatt, L., Németh, A.H., Meesaq, A. et al. Autosomal recessive spinocerebellar ataxia and peripheral neuropathy with raised α-fetoprotein. J Neurol 251, 805–812 (2004). https://doi.org/10.1007/s00415-004-0427-y
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00415-004-0427-y