
Abstract
Cation–π interactions are known to be one of the strongest noncovalent forces in the gas phase, but they rarely occur in a fully solvated environment. The present work used two different ab initio molecular dynamics-based approaches to describe the correlation between the strength of the cation–π interactions and the number of water molecules surrounding the cation. Five different complexes between an aluminum cation and different molecules containing aromatic rings were studied, and the degree of hydration of each complex was varied. Results indicated that cation–π interactions vanish when the aluminum cation is surrounded by more than three water molecules. The results also highlighted the influence of –OH ligands on the interaction strength.

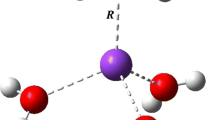
Visualization of the cation–π interaction between the aromatic ring in phenylalanine and the Al3+ cation, together with the corresponding Wannier function centers






Similar content being viewed by others


References
Zatta P, Lucchini R, van Rensburg SJ, Taylor A (2003) Brain Res Bull 62:15–28
Kawahara M (2005) J Alzheim Dis 8:171–181
Dougherty DA (1996) Science 271:163–168
Ma JC, Dougherty DA (1997) Chem Rev 97:1303–1324
Costanzo F, Valle RGDJ (2008) Phys Chem 112:12783–12789
Larrucea J (2009) Computational study of the effect of aluminum cation on aromatic amino acids (Ph.D. thesis). Euskal Herriko Unibertsitatea UPV/EHU, Donostia
Larrucea J, Rezabal E, Marino T, Russo N, Ugalde JM (2010) J Phys Chem B 114:9017–9022
CPMD Consortium (2001) CPMD v3.11.1, C. (revision A11). IBM Corporation/Max-Planck Institut, Stuttgart. http://www.cpmd.org
Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865–3868
Vanderbilt D (1990) Phys Rev B 41:7892–7895
Laasonen K, Car R, Lee C, Vanderbilt D (1991) Phys Rev B 43:6796–6799
Laasonen K, Pasquarello A, Car R, Lee C, Vanderbilt D (1993) Phys Rev B 47:10142–10153
Car R, Parrinello M (1985) Phys Rev Lett 55:2471–2474
Nosé SJ (1984) Chem Phys 81:511
Hoover WG (1985) Phys Rev A 31:1695
Evans DJ, Holian BLJ (1985) Chem Phys 83:4069
Sprik M, Ciccotti GJ (1998) Chem Phys 109:7737–7744
Ciccotti G, Kaprai R, Vanden-Eijnden E (2006) Chem Phys Chem 6:1809
Dunbar RC, Klippenstein SJ, Hrusak J, Stoeckigt D, Schwarz H (1996) J Am Chem Soc 118:5277
Larrucea J (2011) Phys Scr 84:045305
Suipizi M, Carloni PJ (2000) Phys Chem B 104:10087
Swaddle TW, Rosenqvist J, Yu P, Bylaska E, Philips BL, Casey WH (2005) Science 308:1450–1453
Takashi Ikeda MH, Kimura TJ (2006) Chem Phys 124:074503–1
Bock CW, Markham GD, Katz AK, Glusker JP (2006) Theor Chem Acc 115:100–112
Sillanpää A, Päivärinta JT, Hotokka MJ, Rosenholm JB, Laasonen KJ (2001) Phys Chem 105:10111–10122
Acknowledgments
This research was mostly funded by Euskal Herriko Unibertsitatea (the University of the Basque Country), Gipuzkoako Foru Aldundia (the Provincial Government of Gipuzkoa), and Eusko Jaurlaritza (the Basque Government).
The calculations were performed using the Mare Nostrum supercomputer (PowerPC 970MP) at the Barcelona Supercomputing Center (Centro Nacional de Supercomputación), Juropa (Intel Xeon 5570) at the Jülich Supercomputing Center, and Arina (Itanium II) at the SGI/IZO-SGIker at the University of the Basque Country UPV/EHU.
I wish to acknowledge Prof. Jesus M. Ugalde and many people in NSC Jyväskylä, such as Dr. Jaakko Akola, Prof. Hannu Häkkinen, Prof. Robert van Leuwen, and Oleg O. Kit, for discussions and support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Larrucea, J. Solvent effect on cation–π interactions with Al3+ . J Mol Model 18, 4349–4354 (2012). https://doi.org/10.1007/s00894-012-1433-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-012-1433-0