
Abstract
Atopic dermatitis (AD) is the most common chronic inflammatory skin disease in the world. AD is a complex pathology mainly characterized by an impaired skin barrier, immune response dysfunction, and unbalanced skin microbiota. Moreover, AD patients exhibit an increased risk of developing bacterial and viral infections. One of the most current, and potentially life-threatening, viral infection is caused by herpes simplex virus (HSV), which occurs in about 3% of AD patients under the name of eczema herpeticum (EH). Following a first part dedicated to the clinical features, virological diagnosis, and current treatments of EH, this review will focus on the description of the pathophysiology and, more particularly, the presently known predisposing factors to herpetic complications in AD patients. These factors include those related to impairment of the skin barrier such as deficit in filaggrin and anomalies in tight and adherens junctions. In addition, low production of the antimicrobial peptides cathelicidin LL-37 and human β-defensins; overexpression of cytokines such as interleukin (IL)-4, IL-13, IL-25, IL-33, and thymic stromal lymphopoietin (TSLP); or downregulation of type I to III interferons as well as defect in functions of immune cells such as dendritic, natural killer, and regulatory T cells have been involved. Otherwise, genetic polymorphisms and AD topical calcineurin inhibitor treatments have been associated with an increased risk of EH. Finally, dysbiosis of skin microbiota characterized in AD patients by Staphylococcus aureus colonization and toxin secretion, such as α-toxin, has been described as promoting HSV replication and could therefore contribute to EH.
Similar content being viewed by others

Introduction
Atopic dermatitis (AD) is the most common inflammatory skin disease, impacting approximately 15 to 25% of children and 1 to 5% of adults worldwide, with a greater frequency in recent years in industrialized countries, and it is associated with high socioeconomic costs [1,2,3]. In addition to the discomfort caused by symptoms of itching and soreness, adverse consequences on patients’ quality of life have been clearly identified, resulting in sleeplessness, decreased academic and professional performances, loss of self-esteem, and increased family stress [4, 5].
AD is characterized by intense pruritus and skin dryness associated with cutaneous hyper-reactivity to environmental stimuli such as allergens, irritants, food, and humidity as well as stress, viral infections, and bacterial infections [6]. Although AD pathogenesis is not fully understood, complex interactions between skin barrier defect, immune response, skin microbiota, and environmental exposures have been suggested to play key roles [1,2,3, 7, 8]. During evolution of the disease, innate and adaptive immunities are intimately linked and their cross-talks have a clearly established role in the development, maintenance, and flare-up of AD [1].
The main identified critical factors in AD are impaired skin barrier; overexpression of Th2 cytokines, especially interleukin (IL)-4, IL-13, and epithelial cell–derived cytokines IL-25, IL-33, and thymic stromal lymphopoietin (TSLP); low production of antimicrobial peptides (AMPs) and interferons (IFNs); abnormalities in number and/or activity of immune cells such as dendritic cells (DCs), natural killer (NK), and regulatory T cells (Tregs); and dysbiosis of skin microbiota [1, 2, 6, 7]. All of these factors considerably increase the risk of viral or bacterial infection emergence in patients with AD [9, 10]. These complications include infections by Staphylococcus aureus, herpes simplex virus (eczema herpeticum), vaccinia virus (eczema vaccinatum), molluscum contagiosum virus (eczema molluscatum), or coxsackievirus (eczema coxsackium) [9, 11]. This review aims to describe the clinical features and predisposing factors for eczema herpeticum (EH), a severe viral infection occurring in AD patients.
Eczema Herpeticum: a Rare but Severe Viral Infection in AD
One of the most common viral infections occurring in AD patient is EH: a herpes simplex virus (HSV) skin infection [9, 11, 12]. Although EH is relatively rare, occurring in about 3% of AD patients, this initially local disease may progress to a potentially life-threatening systemic infection [9,10,11,12]. The pathogenesis of EH remains largely unknown so far.
Human Herpes Virus Simplex
HSV is an enveloped double-stranded DNA virus belonging to Herpesviridae family and Alphaherpesvirinae subfamily, including HSV type 1 (HSV-1) and HSV-2, which share almost 50% of homologous sequences [10, 13]. Humans are frequently exposed to these pathogens: seroprevalence for HSV-1 is about 80% for children and up to 90% for adults whereas HSV-2 affects 12–15% of adults [10, 13]. HSV-2 is mainly involved in sexually transmitted genital herpes while HSV-1 usually causes labial, ocular, and primary genital herpes [14]. Transmission of HSV occurs after contact with infected secretions. Virus can spread via viral shedding during symptomatic or asymptomatic recurrent infections [15].
HSV usually infects skin or mucosa and replicates in stratified squamous epithelium [14]. Consequently, keratinocytes, the predominant cell type in the epidermis, are the first cellular targets for HSV in skin [16]. The entry of HSV in cells depends on the interaction between viral glycoproteins B and D with host cell surface molecules such as 3-O-sulfate heparan sulfate, nectin-1, and HSV entry mediator, also known as tumor necrosis factor receptor superfamily member 14 (TNFRSF14) enabling the fusion of host cell and viral membranes [10, 17, 18]. Keratinocytes express all these receptors enabling fast virus entry [14], about 90% of viral particles being internalized at 30 min post-infection [16]. Other cells found in skin such as DCs may also be infected with HSV, especially epidermal resident Langerhans cells (LCs), which express receptors involved in HSV entry [14, 19]. Infection of DCs leads to inhibition of their maturation and consequently to reduction in their cytokine production [19]. Dermal fibroblasts are also potential targets for HSV due mainly to high expression of nectin-1 [20]. In addition, entry receptors for HSV are found in various kinds of cells such as monocytes, B- and T-lymphocytes, and endothelial and mast cells [21]. Following the lytic life cycle, HSV starts a lifelong persistence, called latency, established in the sensory ganglia innervating the site of infection. Reactivation of latent viruses can be induced by many factors, such as ultraviolet exposure, emotional stress, fever, tissue damage, or immune suppression [17, 22]. HSV is known to implement multiple strategies to evade host antiviral innate immune response, a phenomenon contributing to infections [17, 23]. While HSV is usually responsible for harmless infection of cutaneous mucous membranes in healthy patients, a disseminated HSV infection can occur on lesional skin of AD patients.
Characteristics of EH
EH was first described in 1887 by Moritz Kaposi, an Austrian dermatologist, in ten children with an “eczema larvare infantum” displaying a vesiculopustular eruption called “Kaposi’s varicelliform eruption” [10,11,12, 15]. Today, this term is applied to all viral complications caused by HSV in skin diseases such as AD, psoriasis, and rosacea. However, the term “eczema herpeticum” is restricted to HSV extensive infection appearing in AD [10,11,12]. EH can be classified according to its severity: from local to disseminated infection, at times leading potentially life-threatening to herpetic encephalitis [12, 15]. Inevitably, EH increases the cost of care for AD patients. For example, in the United States of America (USA), the total cost of inpatient care for EH is about $ 2,000,000 US dollars per year [24].
Epidemiology
In more than 90% of cases, EH is caused by HSV-1, HSV-2 being preferentially located in the genital area [25]. This disease is related not only to primary infection, but also to reactivation of the latent virus [11, 26]. Nonetheless, even if a wide range of person is HSV-seropositive, EH rarely occurs in AD patients. Concerning EH prevalence, about 3% of patients with AD are estimated to develop EH (ADEH+) during their lifetime [27]. In the USA, there were between 4.03 and 7.3 children per million children per year hospitalized for EH between 2002 and 2012 [24]. ADEH+ patients are usually younger than AD patients without a history of EH (ADEH−) [25, 26] and more often male [24, 25, 28]. They display early-onset AD and a higher average scoring atopic dermatitis (SCORAD) index [25, 26, 28]. Risk factors of EH in AD patients include presence of other allergic diseases such as food allergy and asthma [25, 26], lymphopenia with normal white blood cell count, increased erythrocyte sedimentation rate [26], and higher levels of immunoglobulin (Ig) E and circulating eosinophils [25, 26, 28]. EH may also be associated with psoriasis, asthma, or immune suppression [24]. In addition, atopy to a wide diversity of allergens such as Staphylococcus aureus toxins, animal dander, food allergens, house dust, or grasses is noted in ADEH+ patients [25]. ADEH+ patients are also more frequently concerned by S. aureus infections that could lead to septicemia [15, 25, 28]. Finally, EH is independent of season and of time of the year [24, 26]. The predisposing cellular factors for EH are presented in a dedicated section.
Clinical Presentation
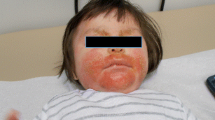
Classically, EH most frequently affects the face, neck, and trunk even if the hands, legs, or genital organs may also be involved (Fig. 1; [29,30,31,32,33,34,35,36,37,38,39,40]). Typical lesions of EH are monomorphic eruptions of dome-shaped papulovesicles (Fig. 1; [10,11,12]). After 1 or 2 weeks, blisters dry and form crusts that fill eroded pits [11, 12]. Vesicles may also be umbilicated and the presence of ulcerations or slits is possible [32, 36, 41,42,43,44,45]. Usually, lesions are completely dried and healed after 2 to 6 weeks [10,11,12]. EH is often accompanied with fever, malaise, or lymphadenopathy (Table 1; [10,11,12]). Diagnosis EH is important because functional and/or life-threatening complications may occur. HSV kerato-conjunctivitis or keratitis may be responsible for blindness [29, 46]. Similarly, HSV gingivostomatitis may decrease dietary intake and hydration [47]. In drastic cases of widespread EH, viremia, meningitis, or encephalitis can be noted and prove fatal [11, 15, 48, 49]. In immunocompromised patients, EH can cause severe hepatitis that may progress to fulminant or acute liver failure [50]. Pregnancy is also a period of heightened susceptibility to HSV infection with an increased risk of dissemination, EH becoming potentially dangerous for the mother and the fetus through materno-fetal viral transmission and subsequent neonatal HSV infection [31, 38, 42]. One of the other main complications of EH is a potential secondary infection of vesicles by bacteria as S. aureus leading to pustules [10, 15, 51]. Similarly, septicemia due to S. aureus superinfections can cause multiorgan failure and can result in the death of the patient [15, 52, 53]. In some cases, clinical manifestations of EH can be atypical, complicating and delaying diagnosis, and threatening the patient’s life. Clinical presentations without emergence of vesicles [30] or with only small papules have been described [54]. Those cases exemplify the diversity of clinical characteristics complicating EH diagnosis. Moreover, differential diagnose may appear, including impetigo, eczema vaccinatum (EV), contact dermatitis, and chickenpox [11, 12, 15, 33, 53] requiring differential management. The difficulty of establishing diagnosis renders it important clinicians should pay attention to any deterioration in an AD patient.

Characteristic monomorphic eruptions of eczema herpeticum on arm (a) and hand (b), and widespread eruption of EH (c)
Diagnosis
Diagnosis of EH is mainly clinical [11, 15, 33, 51, 53]. However, due to the potential diversity of EH clinical manifestations, physicians confirm their diagnosis using diverse laboratory techniques such as Tzanck smear, HSV culture, direct immunofluorescence (IF), or polymerase chain reaction (PCR) (Table 1; [11, 33, 51, 53]). All these techniques present advantages and drawbacks, particularly regarding sensitivity and specificity [33, 51]. Nowadays, molecular biological techniques are preferentially chosen because of their ease of use, reliability, and high sensitivity [55]. Indeed, PCR is positive in 100% of cases when performed on early lesions, and positivity rates remain higher than 80% on later lesions as well (over 30 days) [55]. Because of potential severe complications and fatal outcomes, diagnosis must be established without delay, upon the first consultation. Treatment should start based on clinical characteristics as soon as the infection is suspected without waiting for the results of the complementary analyses [15, 31, 33, 56].
Treatments
Antiviral therapy should be set up as soon as EH is suspected, because in case of severe form, EH can potentially be fatal with a mortality rate of 10 to 75% in the absence of antiviral treatment [11, 12, 57]. Acyclovir (ACV) is the first-line drugs for treatment of HSV infections since the 1980s. This molecule is a guanosine analogue which must be triphosphorylated, firstly by the virus-encoded thymidine kinase (TK) and subsequently by cellular enzymes, to be efficient. Then, ACV-triphosphate acts as a potent inhibitor of viral DNA polymerase [58]. Five to 10 mg/kg of intravenous ACV, 3 times daily, for 5 to 7 days is the recommended treatment for EH [11, 15]. As ACV has poor oral bioavailability, valacyclovir (VACV), an l-valyl-ester prodrug of ACV, has been developed [58, 59]. In treatment with VACV, 500 mg orally twice a day for 5 days is recommended for EH [15]. Penciclovir (PCV) and its oral prodrug famciclovir act similarly to ACV and may be used in countries where it is marketed in dosages of 5 mg/kg/dose and 500 mg, twice a day for 7 days [11, 58, 59].
Sometimes, these treatments can fail due to drug resistance [11]. In a cohort of 8 ADEH+ patients, 3 had an acyclovir-resistant HSV [60]. This resistance may be conferred by viral mutations either in the unique long (UL) 23 gene which encodes the TK in 95% of cases or in the UL30 gene which encodes the viral target DNA polymerase in the 5% of remaining cases [58, 61]. In order to counteract this resistance, mainly supported by TK mutations, other treatments can be used such as foscarnet (FOS) and cidofovir (CDV). FOS and CDV are a pyrophosphate analogue and an acyclic nucleoside monophosphate, respectively, which directly inhibit the viral DNA polymerase without the need for a functional TK. Forty mg/kg 3 times a day for 2 to 3 weeks are recommended for foscarnet [15]. However, both are nephrotoxic and have very poor oral bioavailability; as a result, they are used only when other therapies have failed [12, 51, 58, 59, 61]. These antiviral treatments are not used for topical applications, except in cases of herpetic keratitis where topical antiviral ointment must be applied [12]. Topical glucocorticosteroids may be added to the antiviral drug even if they remain controversial [11, 12]. In addition to a curative approach, a prophylactic treatment should be implemented for ADEH+ patients with high risk of recurrences using oral ACV in a dosage of 200 mg 3 times a day or VACV helping to significantly decrease disease severity [11, 51, 56].
In addition to these classical treatments, new therapeutic approaches may be considered especially in patients infected with a resistant strain. Among these, the IFN-γ therapy may be a good alternative in infection with resistant strains, particularly for AD patients with a low level of IFN-γ [51, 60]. Indeed, IFN-γ enables control of HSV replication and propagation, and may be of considerable benefit in cases of EH. Nevertheless, up until now, patients’ clinical improvement is not sufficient, given the very high cost of this therapy [12, 51, 60].
Predisposing Factors for Eczema Herpeticum in Atopic Dermatitis Patients
Impairment of the Skin Barrier
AD is characterized by impaired barrier function which cause increased transepidermal water loss and may facilitate entry of HSV through the first keratinized layers of the skin and binding to its receptors at the surface of the target cells [2, 11]. This dysfunction arises from a defect in terminal keratinocyte differentiation due to decreased expression of filaggrin (FLG) that damages the structure and composition of the stratum corneum. In addition, anomalies in tight junctions (TJs) and adherens junctions (AJs) are involved in this process (Fig. 2).

Factors promoting HSV replication in eczema herpeticum. Atopic dermatitis (AD) patients are characterized by impaired skin barrier, especially a filaggrin and claudin deficit as well as increased Nectin-1 accessibility. An abnormal immune response is found and includes overexpression of type 2 cytokines such as interleukin (IL)-4 and IL-13 and other inflammatory mediators such as IL-25, IL-22, and TSLP. These cytokines lead to lower induction of antimicrobial peptides than in other inflammatory skin diseases and exacerbate skin barrier defects. During the development of EH in AD patients (ADEH+ patients), immune cells are also affected with a lack of plasmacytoid dendritic cells (pDCs), defective natural killer cells (NK), expansion of regulatory T cells (Treg), and overexpression of IDO1 by DCs altering the production of interferon (IFN), an important antiviral cytokine. Finally, the lesional skin microbiota of AD patients is unbalanced with sizable colonization of S. aureus producing toxins such α-toxin. All these modifications interact with each other, promoting entry or reactivation and replication of HSV and, finally, the development of EH
Filaggrin
FLG is a key protein of the barrier function, playing an important role in formation of the cornified cell envelope [62, 63]. Proteolytic degradation products of FLG are involved in maintenance of skin hydration and the pH of the skin, and play a crucial role in the regulation of key biochemical events, including protease activity, desquamation, lipid synthesis, barrier permeability, and cutaneous antimicrobial defense [7, 63, 64]. However, in AD patients, FLG expression is strongly reduced. This lack of FLG can be explained, in 1/3 of patients, by loss-of-function mutations in the gene encoding FLG [62]. Two common mutations, R501X and 2282de14, are associated with AD (Table 2; [64]). R501X mutation occurs in 18% of AD patients and 2282del4 in 48% [64, 65]. Gao et al. have studied those mutations in 278 Euro-American AD patients including 112 ADEH+ patients [65]. They found that 25.4% of ADEH+ patients carried the R501X mutation and 17.8% the 2282el4 one. Moreover, a significant association for the R501X mutation in ADEH+ patients was found. In addition, ADEH− patients carrying both R501x and 2282de14 mutations have an increased risk of ADEH compared to those with only one mutation [65].
Nevertheless, less than 1/3 of ADEH− patients are concerned by these mutations, which is why other mechanisms, including cytokine environment of AD lesions (detailed later in a dedicated paragraph), are likely to contribute to epidermal barrier defects and development of viral infections [7, 62, 63, 72]. In addition, the reduced level of FLG results in decreased formation of FLG breakdown products like urocanic acid (UCA) and pyrrolidone carboxylic acid (PCA), which help to maintain acid pH on the skin [7, 63, 64, 72]. Indeed, incubation of keratinocytes with UCA and PCA at various concentrations has caused dose-dependent medium acidification and allowed significant dose-dependent inhibition of HSV-1 replication [72]. Viral replication was also reduced with a decreasing pH due to hydrochloric acid [72] suggesting that an acid pH inhibits the entry of HSV into cells [72, 73]. Consequently, reduction of UCA and PCA due to FLG deficit causes elevation of the skin-surface pH, conducive to HSV-1 replication that can increase the risk of EH [72].
Tight Junctions
TJs, which are complex of adhesive proteins, control the entry of water and solutes across the skin and establish a link between keratinocytes within the stratum granulosum [27, 63]. Healthy TJs prevent keratinocytes from viral propagation by contact between cells blocking access to the virus receptor [66]. TJs are composed of three main transmembrane proteins: claudin (CLDN) which is involved in TJs resistance and permeability, occludin, and the IgG-like family of junctional adhesion molecules (JAMs) [63, 74]. Gene expression profiling in AD patients shows a reduction in CLDN1 and CLDN23 expressions [67]. In addition, lower electrical resistance and absence of ion selectivity permeability are signs of TJ defects in AD patients [67]. Defect in CLDN1 can be partially explained by an adjacent single-nucleotide polymorphism (SNP) (rs9290927) in the gene encoding CLDN1 associated with a higher risk of AD and two SNPs (rs893051 and rs9290929) associated with disease severity [67]. As described for FLG and after exclusion of FLG mutations, an intronic SNP (rs3732923) and a SNP in promoter region (rs3954259), in Euro-American and Afro-American populations, respectively, seem significantly associated with EH (Table 2; [66]). The role of CLDN1 in HSV infection has been studied using CLDN1 siRNA transfected primary human keratinocytes infected with a virulent HSV-1 strain [66]. A significant increase in the number and size of HSV-1 focal forming units was observed in CLDN1 siRNA transfected keratinocytes in comparison to control siRNA transfected keratinocytes [66]. These results suggest that a lower CLDN1 level may promote HSV infection and the spread of the virus from one keratinocyte to another [66]. In another study, the role of TJs in HSV-1 entry was investigated using either murine primary keratinocytes deficient for the polarity regulator partitioning defective-3 (Par3) or knockout keratinocytes for E-cadherin [75]. Deficit of Par3 and E-cadherin led to non-functional TJs and a reduced transepithelial electrical resistance [75]. In both cases, an increasing number of cells expressing HSV antigens was observed compared to control cultures. These results support the fact that defects in TJs facilitate virus access to host cell receptors, penetration in the skin, and, finally, HSV infections [75].
Adherens Junctions
AJs are formed by the nectin-afadin complex including nectin-1 to nectin-4 and the cadherin-catenin complex such as E-cadherin and β-catenin, and have a structural intercellular adhesive role [74]. Moreover, nectin-1 is a HSV-glycoprotein D receptor, allowing the entry of HSV into cells [18]. It has been shown that when AJs are disrupted, nectin-1 is detectable on cell surfaces that increase its availability [76]. Consequently, disruption of AJs increases the availability of nectin-1, which becomes more accessible for HSV, and facilitate virus entry into cells [76]. An alteration of AJs in AD patients might occur and eventually be considered a risk factor for EH.
Altered Antimicrobial Peptide Production
When the epidermal barrier is disrupted, activation of the skin innate immune system prevents the entry of pathogens in the organism. Among the effectors of innate immunity, AMPs are able not only to directly kill bacteria or viruses but also to activate adaptive immunity [77, 78]. In human skin, different AMPs contributing to antimicrobial defenses are produced: the cathelicidin LL-37, the human β-defensins (hBD) 1–4, the psoriasin (S100A7), and the ribonuclease 7 (RNase 7) [77, 78]. Some are constitutively expressed in epithelial tissue such as LL-37, hBD-1, S100A7, and RNase 7, whereas hBD-2, hBD-3, and hBD-4 are present at low levels in normal skin but can be strongly induced in infectious or inflammatory conditions [77, 78]. β-defensins and LL-37 are known to exert an antiviral activity against HSV [27, 79].
In inflamed skin of AD patients, levels of hBD-2, hBD-3, LL-37, Rnase 7, and S100A7 are slightly higher than those in healthy skin [78]. Nevertheless, their expression and production except for RNase 7 are clearly lower in the skin of AD patients as compared to psoriatic skin [78, 80, 81]. These low AMP levels in ADEH− patients may confer a higher risk for development of bacterial and viral infections such as EH (Fig. 2; [10, 80]).
LL-37
Previously, it has been shown that LL-37 contributes to control HSV skin infections. In human keratinocytes infected with HSV-2 and then treated with exogenous LL-37, viral replication has been significantly reduced [82]. In HSV-1-infected primary keratinocytes, the anti-HSV-1 activity of LL-37 appeared to be due to the potentiation of cellular antiviral defenses through the induction of interferon-stimulated gene expression [79]. In addition, the role of cathelicidin in HSV infections has been tested in knockout (KO) mice for the gene encoding murine cathelicidin. In skin biopsies collected from these mice, HSV-2 replication was significantly higher than that in skin biopsies from wild-type (WT) mice suggesting that cathelicidin is involved in protection against HSV skin infections [82]. Level of LL-37 in ADEH+ patients in comparison with ADEH− patients has been studied using immunostaining of biopsies from lesional skin [82]. A lower level of LL-37 in ADEH+ patients compared to ADEH− patients has been observed suggesting that ADEH+ patients present exacerbated cathelicidin deficit that could facilitate HSV replication [82]. In addition, a significant negative correlation between cathelicidin expression and total serum IgE has been found in AD patients suggesting that marked Th2 polarity reduces LL-37 induction during inflammation [82, 83]. In this way, stronger reduction in skin cathelicidin expression may predispose patients with AD to develop EH [82].
Human β-Defensins
Defensins seem to have an antiviral activity against HSV through direct binding to the viral envelope glycoproteins [11, 77]. In addition, hBD-3 is able to bind host cell receptors, thereby avoiding virus-host cell interactions and preventing HSV attachment [84, 85]. In lesional skin biopsies, lower expression of hBD-2 and hBD-3 has been found in ADEH+ patients than in ADEH− patients [83]. As for LL-37, Th2 cytokines, especially IL-4 and IL-13 overexpression in AD, contributes to the downregulation of hBD-3 in keratinocytes [81]. In lesions of ADEH+ patients, the levels of these cytokines were higher than those in ADEH− patients, which may explain the exacerbated hBD-3 deficit [83]. Consequently, defensins deficit in AD patients may also be a risk factor for the development of HSV infections.
Cytokine Environment
In AD skin lesions, T cell and dendritic cell infiltrates appear to trigger the production of cytokines [1]. The cytokine profile of lesional skin is complex and depends on the phase of the disease and the phenotypes of AD patients, which are heterogeneous [6]. AD is characterized by different types of immune responses that can occur concomitantly. A Th2/Th22 polarization is predominant, covering the whole disease spectrum with considerable cytokine activation and production during acute disease [86,87,88]. This immune response is constituted of Th2 (IL-4, IL-13, IL-31) and Th22 (IL-22) cytokines [6, 87, 88]. In chronic AD lesions, an increase of the Th1 immune response appears [86, 87, 89]. An attenuated Th17 response and IL-17 production as compared to other inflammatory diseases such as psoriasis is also found in chronic and acute AD [1, 86,87,88]. In addition, other mediators such as IL-25, IL-33, and TSLP, which are known to promote Th2 responses, are overexpressed in AD patients [6, 7, 90]. In contrast, IFN-γ and type I IFNs, which have a crucial role in the antiviral defense, are downregulated in AD patients [7, 9, 91]. All these modifications may contribute to the development of EH in AD patients (Fig. 2).
IL-4, IL-13
As evocated in previous sections, IL-4 and IL-13, which are strongly expressed in AD, can promote viral infections and EH development due to their contribution to FLG and AMP expression deficit [62, 72, 81, 82]. Indeed, addition of IL-4 and IL-13 to keratinocytes was shown to downregulate significantly filaggrin [62, 72] and hBD-3 expression [81]. These cytokines play also a role in viral replication: in IL-4- and IL-13-pretreated keratinocytes, HSV-1 gene expression considerably increased 24 h post-infection [72]. IL-4 and IL-13 signaling pathways involve the signal transducer and activator of transcription 6 (STAT-6). Genotyping of ten STAT-6 SNPs in Euro-American populations has shown a significant association of three intronic STAT-6 SNPs with ADEH: i.e., rs3024975, rs841718, rs167769, and one ′UTR SNP, rs703817 (Table 2; [68]). Two SNPs, rs3024951 and rs324013, were associated with differential IFN-γ production in HSV-stimulated peripheral blood mononuclear cells (PBMCs) isolated from ADEH+ patients. Rs324013 was simultaneously associated with an increased risk of ADEH+ and a reduced level of IFN-γ [68]. These results suggest involvement of STAT-6 SNPs in the regulation of IFN-γ production, leading to the development of EH. Finally, characterization of virus-specific T cell responses has revealed increased of IL-4 secretion in HSV-1-stimulated cells from ADEH+ patients compared to healthy and ADEH− patients corresponding to higher Th2 polarization in ADEH+ patients than in ADEH− patients [92]. Consequently, virus-specific T cells display a Th2 immune response that is exacerbated in ADEH+ patients which may contribute to development of viral infections [92].
IL-25
IL-25 is an IL-17 cytokine produced by keratinocytes and mononuclear cells, overexpressed in AD patients compared to healthy patients [93]. In addition to its role in the stimulation of Th2 differentiation, IL-25 is involved in the downregulation of FLG expression in keratinocytes alone or in synergy with IL-4 and IL-13 [72, 93]. IL-25 treatment prior to HSV-1 infection has led to a significant increase of viral replication in comparison to untreated cells [72]. This effect was accentuated when keratinocytes were pretreated with a combination of IL-25, IL-4, and IL-13. These findings suggest that IL-25, conjointly with IL-4 and IL-13, can promote HSV-1 replication in AD patients [72]. However, contrary to IL-4 and IL-13, IL-25 did not inhibit LL-37 and hBD-3 expression in HSV-1-infected keratinocytes, thereby highlighting a proviral effect independent of AMP expression [72]. In FLG siRNA–treated keratinocytes, no difference in HSV-1 gene expression was noted in presence or absence of IL-25 suggesting that IL-25 enhances HSV-1 replication only by inhibiting FLG expression [72]. Finally, IL-25 levels were found to be higher in skin biopsies of ADEH+ patients compared to ADEH− or healthy patients, suggesting that overexpression of IL-25 may also be considered a predisposing factor to EH [72].
TSLP
TSLP, an IL-7-like cytokine, is known to target DCs in order to induce Th2 cell differentiation and to interact with skin-homing Th2 cells enhancing IL-4 production [69, 94]. TSLP acts by binding the IL-7 receptor α-chain (IL-7Rα) and the TSLP receptor chain (TSLPR) [69, 94]. In AD patients, TSLPR expression in CD4+ T cells was higher than that in healthy and psoriatic patients [94]. In addition, it was shown that treatment of CD4+ T cells with IL-4 increased TSLPR expression and that stimulation of CD4+ T cells from ADEH− patients with TSLP increased production of IL-4 by about 30% [94]. Consequently, a feedback loop mechanism between TSLP and IL-4 may be involved in AD through potentiation of Th2 profile. Moreover, genotyping of 29 SNPs of TSLP, IL-7R, and TSLPR in Euro-American population has shown that two TSLP SNPs, rs1898671 and rs2416259, were significantly associated with ADEH+ as well as four IL-7R SNPs namely rs12516866, rs10213865, rs1389832, and rs10058453 (Table 2; [69]). Interestingly, TSLPR SNPs were associated with ADEH− but not necessarily with ADEH+ [69]. TSLP SNP rs1898671 was even associated with a lower risk of ADEH+ [69]. These results suggest that TSLP signaling pathway may be involved in EH. Moreover, in a mouse model of EV, it was shown that in both primary and satellite lesions, VV dissemination to internal organs and induction of skin inflammation were inhibited in TSLPR-deficient mice [95]. Similar results were obtained in mice depleted for IL-33 receptor [95] suggesting that increased levels of TSLP and IL-33 in AD patients can contribute to the development of viral infections.
IFNs
IFNs play an important role in immune responses, activating antigen-presenting cells such as DCs, macrophages, and T and natural killer cells and promoting differentiation of T helper cells into Th1 cells [27, 70, 96, 97]. Following activation of type I IFN by viruses, transcription of numerous interferon-stimulated genes such as MX1, OAS1, BST2, and PKR, involved in cell antiviral defenses, is initiated [98]. IFNs play an antiviral effect against HSV-1 inhibiting viral replication [97]. Mikloska et al. have shown a reduction of HSV-1 titer of about respectively 60%, 50%, and 90% using plaque reduction assay, in skin explants preincubated with IFN-α, IFN-γ, or both before infection [99]. In another study, pretreatment of keratinocytes with IFN-γ significantly decreased HSV-1 replication, thereby confirming the major role of IFN-γ in the antiviral defenses [72]. ADEH+ patients are characterized by a deficit in antiviral IFN response [9,10,11, 71, 92]. Indeed, ADEH+ patients’ T cells stimulated with HSV-1 disclosed a significant reduction in the IFN-γ response compared to those from healthy patients [71, 92]. Moreover, the IFN-γ response reduction seemed to be more accentuated in ADEH+ patients than in ADEH− patients [92]. In addition, the number of IFN-γ producing cells was lower in ADEH+ patients in comparison with ADEH− and healthy patients [92]. The lower expression of IFNs in EH may result from different causes. First, transcriptomic analysis performed on HSV-1-infected PBMCs revealed pronounced downregulation of 15 types I and III IFN genes including IFN-α and type III IFN IL-29, in ADEH+ patients compared to ADEH− patients [100]. Then, significant downregulation of IFN (α, β, and ω) receptor I, IFN-γ, and IFN-γ receptors was also found in ADEH+ patients [71].
Furthermore, the expression of interferon regulatory factors (IRF) 2, 3, and 7, transcription factors regulating IFN expression and cellular antiviral response, was reduced in HSV-1-stimulated PBMCs from ADEH+ patients compared to ADEH− and healthy patients [100] and IRF2 gene expression was reduced in skin biopsies of ADEH+ patients compared to ADEH− and healthy patients [70]. IRF3 and IRF7 pathways involve ankyrin repeat domain 1 (ANKRD1) as a binding partner [101] and it has been reported that silencing ANKRD1 in DCs caused a significant decrease in IL-29 and IFN-β gene expression, suggesting involvement of ANKRD1 in the type I and III IFN pathways especially in the IRF3 pathway [101]. However, ANKRD1 expression in HSV-1-infected PBMCs was lower in ADEH+ patients compared to ADEH− patients suggesting that this downregulation may contribute to reduced IFN levels in ADEH+ patients [101]. Otherwise, the association between IFN-γ and IFN-γ receptor 1 gene variants and ADEH+ patients has been also tested (Table 2; [71]) showing that two IFN-γ SNPs, rs2069727 and rs2430561, were significantly associated with lower IFN-γ production following HSV infection [71]. In addition, IFN-γ receptor 1 SNPs, rs1327475 and rs10457655, had a stronger association with ADEH+ and may increase the risk of developing viral infections [71]. Finally, genetic variants of IRF2 found in North American populations should also be considered a risk factor [70] as several IRF2 SNPs such as rs3775543, rs377552, rs809909, rs7655371, rs6812958, rs2797507, rs11132242, and rs17488073 were associated with an increased risk of ADEH+ (Table 2; [70]).
On the other hand, a large amount of IFN-α and IFN-β may be produced by plasmacytoid DCs (pDCs) in viral infection [102]. However, flow cytometry analyses have shown that pDCs are almost missing in AD lesions [102]. The lack of pDCs, potentially worsened by Th2 cytokines in ADEH− patients, could impair the production of IFN-α and IFN-β and, finally, may predispose to EH [10, 102]. Moreover, increased mRNA level of the suppressor of cytokine signaling-1 (SOCS-1), a protein which negatively regulates IFN activity, has been shown in HSV-1-infected keratinocytes and represents a mechanism of viral immune evasion [103]. Unlike in skin biopsies from psoriasis patients, where an elevated IL-29 expression has been found, skin biopsies from AD patients did not express IL-29 [104]. As IL-29 has been shown to be involved in the expression of antiviral proteins such as MX1, BST2, ISG15, and OAS2, this finding could explain the low expression of these proteins in AD patients [104]. Finally, in keratinocytes and reconstructed epidermis infected by HSV-1, addition of IL-29 decreased infection in a dose-dependent manner [104]. Taken together, these studies indicate that IFN deficits in AD patients reduce antiviral defense and may be considered a risk factor for the development of EH.
Defects in Immune Cells
Various immune cells, such as LCs, NK cells, DCs, T cells, mast cells, eosinophils, and macrophages, are found in inflammatory skin of ADEH− patients [7] but some of their functions are disrupted [91]. This is the case with the inflammatory DCs and LCs present in lesional skin, which negatively regulate T cell functions, thus contributing to lower antiviral defenses [2, 7, 105]. Moreover, number of NK cells is reduced compared to healthy patients and remaining cells are functionally defective [91]. Finally, in skin lesions of ADEH− patients, the number of Tregs is increased [2]. All these dysfunctions found in AD patients may be considered predisposing factors to EH (Fig. 2).
Dendritic Cells
DCs are antigen-presenting cells involved in primary and secondary adaptive immune responses. Skin DCs include LCs, pDCs, and inflammatory epidermal DCs that are important in antiviral defense mechanisms [91, 102, 105]. In addition to their role with regard to T cell function regulation and cytokine production, DCs contribute to the production of indoleamine 2,3-dioxygenase (IDO), a catabolizing enzyme of tryptophan (TRP) in the kynurenine pathway. At low level, the IDO activity has a crucial role during viral infection with an ability to inhibit HSV-2 replication [106]. Nevertheless, high IDO1 activity degrades TRP to a critical level in T cells, leading to an inhibition of their proliferation and consequently a reduced production of IFN-γ [105, 106]. In ADEH+ patients, IDO activity in serum was significantly higher than that in serum of ADEH− and healthy patients [105]. IDO1 can be produced by different cells such as pDCs, monocytes, and LCs. Flow cytometry analyses performed on each of these cells have identified mature LCs as the major source of IDO-1 in ADEH+ patients [105]. This overexpression of IDO1 may be caused by viruses. LCs stimulated with Poly I:C to mimic viral infection have exhibited increased IDO1 expression and activity, a phenomenon exacerbated in LCs from ADEH+ patients compared to those from ADEH− and healthy patients [105]. This result suggests that exacerbated IDO1 expression and activity in LCs due to HSV exposure might be involved in the lack of IFN-γ and might finally function as a predictive biomarker of viral complications in AD [105].
Natural Killer Cells
NK cells, actors of innate immune system, represent about 10–15% of peripheral blood lymphocytes and are major actors in the defense against HSV infections due to their ability to lyse infected cells by producing secretory lysosomes containing cytotoxic proteins, especially granzyme B and perforin [91, 107]. NK cells depleted mice infected with HSV-1 presented earlier and more severe skin lesions and higher mortality rate compared to WT mice, underlining the major role of NK cells in limiting HSV infections [108]. It has been shown that the number of NK cells is significantly reduced in AD patients compared to healthy patients and the reduction was exacerbated in ADEH+ patients [10, 91, 109]. However, in a murine model of EH, no difference in mature cytolytic NK cell number was found between EH mice and WT mice, infected or not with HSV-1 [110]. However, the number of NK cells producing granzyme B, perforin, and IFN-γ was markedly reduced after HSV-1 infection in EH mice compared to WT mice, suggesting that in case of HSV-1 infection, NK cells expressed fewer effector molecules [110]. It has also been shown that transfer of NK cells in EH mice prior to HSV-1 infection reduced the severity of EH [110]. Consequently, defective NK cell activity may increase the EH severity and could be a predisposing factor for EH.
Regulatory T Cells
Regulatory T cells (Tregs), a population of CD4+ T cells, are usually characterized by expression of the alpha-chain of the IL-2 receptor (CD25) and the transcription factor Foxp3 and have an immune suppressive role on effector T cells [2, 111]. It has been shown in HSV-infected mice that a depletion of Tregs may lead to an increase of CD8+ and CD4+ T cell responses, cytotoxicity against HSV, and expression of IFN-γ and granzyme B [111]. This depletion also reduces the viral titer of HSV in brain and lymph nodes [111]. Consequently, Tregs seem to be able to suppress adaptive immune responses established by CD4+ and CD8+ T effector cells against HSV infection. In addition, flow cytometry analyses performed in PBMCs from ADEH+ patients showed that all the Tregs subsets tested were present in a considerably higher concentration than in ADEH− and healthy patients [112]. Moreover, co-culture of Tregs from ADEH+ patients and CD4+ effector T cells caused an inhibition of effector T cell proliferation in comparison with culture of effector T cells alone [112]. Regarding the increase of Tregs frequency and the preservation of normal function, the same results were obtained in EH mice [110]. Consequently, Tregs in ADEH+ patients retained their ability to suppress the activity of effector T cells. A cytokine expression analysis performed on lymphocyte subsets demonstrated an impairment in IFN-γ and TNF-α production by CD8+ T, CD4+ T, and CD56+ NK cells in ADEH+ patients [112]. Then, PBMCs depleted of Tregs and infected with HSV-1 let restore the production of IFN-γ by CD8+ and CD4+ T cells [112]. Taken together, these results suggest that expansion of functional Tregs could promote HSV-1 infection by inhibiting immune responses and be involved in the development of EH.
Consequences of Current AD Therapies on the Occurrence of EH
AD may be managed using different treatments including topical calcineurin inhibitors [6, 113]. Topical calcineurin inhibitors are immunomodulators represented by pimecrolimus, an ascomycin macrolactam derivate, that suppresses production of pro-inflammatory mediators by activated T cells and by tacrolimus, a macrolide molecule that inhibits T cell activation [114, 115]. Study of the efficacy and safety of pimecrolimus (12.4%) showed a slight increase of viral infection in the group of ADEH− patients (with moderate AD) treated with pimecrolimus compared the group of ADEH− patients treated with a conventional treatment control using emollients and topical corticosteroids (6.3%) [116]. In the same study, EH appeared in 2.1% of patients in the group treated with pimecrolimus compared to 0.8% in the control group suggesting a role of pimecrolimus in the development of EH [116]. Similarly, six cases (2%) of EH were reported among 316 patients with moderate to severe AD treated with 0.1% of tacrolimus, a finding suggesting that this drug might be a risk factor for EH [117]. In addition, several clinical cases in the literature have reported EH development during therapies using one of these molecules. For example, two patients with severe AD, in a cohort of 36 patients followed, presented with disseminated herpetic lesions following treatment with tacrolimus [114]. Likewise, after 1 year of treatment with 1% pimecrolimus, patients can develop widespread herpetic vesicles positive for HSV-1 [115]. Overall, topical calcineurin inhibitors are suspected to have a role in the development of viral infections but the available data are insufficient to claim or disprove an association between treatment and an increased risk of EH [114, 115]. Conversely, another study reported that less than 1% of patients treated with tacrolimus ointment monotherapy were diagnosed as having EH [118] and similar results were obtained in a cohort excluding patients with EH history [119]. Despite their beneficial role in the repair of the skin barrier function of AD patients, it cannot be excluded that these immunosuppressive treatments may alter skin antiviral defenses in some cases. In contrast, association between cyclosporin, another immunosuppressor, and infectious diseases in AD has been tested but no significant difference between treated and untreated patients has been found [120]. Lastly, no association between Dupilumab, a monoclonal antibody targeting IL-4 and IL-13 receptor, and EH has been proven [121].
Influence of Microbiota in HSV-1 Infection
An important parameter to take account in EH outbreak concerns skin microbiota. Skin microbiota is composed of a rich community of microorganisms whose abundance and composition change according to individuals, ecologic features of each specific body site, and environment [122]. In spite of possible spatial and temporal fluctuations, four phyla are dominant: Actinobacteria, Proteobacteria, Bacteroidetes, and Firmicutes. Three genders, Staphylococcus (Firmicutes), Corynebacterium (Actinobacteria), and Propionibacterium (Actinobacteria), represent more than 60% of skin-associated bacteria [123, 124]. Commensal bacteria such as Staphylococcus epidermidis and Propionibacterium spp. are essential in normal skin because of their contribution to the regulation of skin immunity, controlling AMP production, neutrophil recruitment, and defense against pathogens [122, 125]. However, AD patients are characterized by a skin microbiota dysbiosis [126].
Dysbiosis of Skin Microbiota in AD Patients
Skin microbiome dysbiosis has been associated with AD and correlated with disease severity [2, 125,126,127]. Sequencing of skin samples from ADEH− and healthy patients showed decreased bacterial diversity in AD lesions [126]. In the antecubital and popliteal creases of ADEH− patients, the genus Staphylococcus represented about 90% of bacteria during untreated flares compared to only 16% of bacteria in healthy patients, 31% in treated flares, and 20% post flare [126]. S. aureus was the most predominant species in AD representing 65% in untreated flares [126]. Thus, dysbiosis of the skin microbiome in AD may have an important role during viral infection and in the emergence of EH. Indeed, it has been shown that a dysbiosis of microbiota led to a more severe HSV-2 infection, thereby illustrating the crucial role of commensal microbiota in viral infections [128]. Moreover, S. aureus is responsible for severe skin infections in ADEH− patients due to the huge amount of virulence factors produced and may be related to an increased risk of viral infections (Fig. 2; [125, 129]).
Role of S. aureus in Viral Infections
Among S. aureus virulence factors, the pore-forming α-toxin is one of the most virulent insofar as it can provoke alteration of cell membrane integrity, activation of stress-signaling pathways, loss of cellular components, modification of ion gradients, and concentration-dependent cell lysis [129, 130]. Interestingly, 24h-treatment of keratinocytes with α-toxin before HSV-1 infection increased viral replication, a phenomenon which was not observed using staphylococcal enterotoxin B or toxic shock syndrome toxin 1 [129]. Co-infection of keratinocytes with α-toxin-deficient strain of S. aureus and HSV-1 resulted in a significantly lower viral load than co-infection with a WT bacterial strain, thereby confirming the role of α-toxin in viral replication [129]. The effect of α-toxin on viral infection seems to depend on the disintegrin and metalloprotease 10 (ADAM10) receptor of α-toxin. Indeed, siRNA silencing of ADAM10 gene expression in keratinocytes has been shown to prevent formation of α-toxin heptamer, with no increase of viral load compared to controls [129]. This result was confirmed using a mutated α-toxin deficient in pore formation, which was not able to enhance HSV-1 replication in keratinocytes [129]. The mechanisms responsible for the enhancement of viral replication by α-toxin did not involve modulation of expression of virus receptor genes, antiviral response genes, or antimicrobial peptide genes [129]. It was shown that α-toxin acts by promoting HSV-1 entry into keratinocytes and consequently contributes to the development of EH in AD patients [129].
Nevertheless, S. aureus produces other virulence factors, especially phenol-soluble modulins (PSMs), which are also able to form pores in cell membrane [125]. One of them, the δ-toxin or PSMγ, has an important role in the physiopathology of AD triggering Th2 response [125, 131]. In addition, δ-toxin was found in a huge amount in AD lesions infected by S. aureus [131]. Similarly, PSMα is able to induce the death of keratinocytes and trigger the production of inflammatory cytokines [132]. Taking into account the role of α-toxin in EH and of PSMs in AD, PSMs might also have a role in viral infection and should be studied in EH.
Conclusion
Because of the different modifications of the skin that occur in AD such as impairment of barrier function, alteration in innate and adaptive immunity, dysbiosis of skin microbiota, and colonization of lesions by S. aureus, AD patients have a higher risk of disseminated viral infections, particularly caused by HSV-1 so-called EH. Indeed, all these modifications promote entry, replication, and propagation of viruses and contribute to the development of EH. EH may lead to dramatic outcomes in case of dissemination throughout the organism, including the death of the patient. This is why it is crucial to diagnose this infection early upon the appearance of the first symptoms and initiate treatment as soon as possible. Nowadays, pathophysiology of EH remains poorly known. While several factors such as cytokine and genetic defects seem to potentiate HSV replication, complementary studies are required to better understand the mechanisms involved. Moreover, HSV seems to interact with pathogens especially S. aureus which is known to amplify viral replication through virulence factors. That much said, many of the virulence factors of S. aureus and its interactions with other bacterial species have yet to be studied. The elucidation of new mechanisms involved in EH may open the way to the development of new therapeutic approaches.
References
Werfel T, Allam JP, Biedermann T, Eyerich K, Gilles S, Guttman-Yassky E, Hoetzenecker W, Knol E, Simon HU, Wollenberg A, Bieber T, Lauener R, Schmid-Grendelmeier P, Traidl-Hoffmann C, Akdis CA (2016) Cellular and molecular immunologic mechanisms in patients with atopic dermatitis. J Allergy Clin Immunol 138(2):336–349. https://doi.org/10.1016/j.jaci.2016.06.010
Bieber T (2008) Atopic dermatitis. N Engl J Med 358(14):1483–1494. https://doi.org/10.1056/NEJMra074081
Nutten S (2015) Atopic dermatitis: global epidemiology and risk factors. Ann Nutr Metab 66(Suppl 1):8–16. https://doi.org/10.1159/000370220
Carroll CL, Balkrishnan R, Feldman SR, Fleischer AB Jr, Manuel JC (2005) The burden of atopic dermatitis: impact on the patient, family, and society. Pediatr Dermatol 22(3):192–199. https://doi.org/10.1111/j.1525-1470.2005.22303.x
Lewis-Jones S (2006) Quality of life and childhood atopic dermatitis: the misery of living with childhood eczema. Int J Clin Pract 60(8):984–992. https://doi.org/10.1111/j.1742-1241.2006.01047.x
Leung DY (2013) New insights into atopic dermatitis: role of skin barrier and immune dysregulation. Allergol Int 62(2):151–161. https://doi.org/10.2332/allergolint.13-RAI-0564
Peng W, Novak N (2015) Pathogenesis of atopic dermatitis. Clin Exp Allergy 45(3):566–574. https://doi.org/10.1111/cea.12495
Elias PM, Hatano Y, Williams ML (2008) Basis for the barrier abnormality in atopic dermatitis: outside-inside-outside pathogenic mechanisms. J Allergy Clin Immunol 121(6):1337–1343. https://doi.org/10.1016/j.jaci.2008.01.022
Ong PY, Leung DY (2016) Bacterial and viral infections in atopic dermatitis: a comprehensive review. Clin Rev Allergy Immunol 51(3):329–337. https://doi.org/10.1007/s12016-016-8548-5
Bussmann C, Peng W-M, Bieber T, Novak N (2008) Molecular pathogenesis and clinical implications of eczema herpeticum. Expert Rev Mol Med 10:e21. https://doi.org/10.1017/s1462399408000756
Wollenberg A, Wetzel S, Burgdorf WH, Haas J (2003) Viral infections in atopic dermatitis pathogenic aspects and clinical management. J Allergy Clin Immunol 112(4):667–674. https://doi.org/10.1016/S0091
Wollenberg A (2012) Eczema herpeticum. Chem Immunol Allergy 96:89–95. https://doi.org/10.1159/000331892
Novak N, Peng WM (2005) Dancing with the enemy: the interplay of herpes simplex virus with dendritic cells. Clin Exp Immunol 142(3):405–410. https://doi.org/10.1111/j.1365-2249.2005.02927.x
Cunningham AL, Diefenbach RJ, Miranda-Saksena M, Bosnjak L, Kim M, Jones C, Douglas MW (2006) The cycle of human herpes simplex virus infection: virus transport and immune control. J Infect Dis 194(Suppl 1):S11–S18. https://doi.org/10.1086/505359
Khan A, Shaw L, Bernatoniene J (2015) Fifteen-minute consultation: eczema herpeticum in a child. Arch Dis Child Educ Pract Ed 100(2):64–68. https://doi.org/10.1136/archdischild-2013-304460
Rahn E, Petermann P, Hsu MJ, Rixon FJ, Knebel-Morsdorf D (2011) Entry pathways of herpes simplex virus type 1 into human keratinocytes are dynamin- and cholesterol-dependent. PLoS One 6(10):e25464. https://doi.org/10.1371/journal.pone.0025464
Suazo PA, Ibanez FJ, Retamal-Diaz AR, Paz-Fiblas MV, Bueno SM, Kalergis AM, Gonzalez PA (2015) Evasion of early antiviral responses by herpes simplex viruses. Mediat Inflamm 2015:593757. https://doi.org/10.1155/2015/593757
Spear PG, Longnecker R (2003) Herpesvirus entry: an update. J Virol 77(19):10179–10185. https://doi.org/10.1128/jvi.77.19.10179-10185.2003
Salio M, Cella M, Suter M, Lanzavecchia A (1999) Inhibition of dendritic cell maturation by herpes simplex virus. Eur J Immunol 29(10):3245–3253. https://doi.org/10.1002/(SICI)1521-4141(199910)29:10<3245::AID-IMMU3245>3.0.CO;2-X
Thier K, Mockel M, Palitzsch K, Dohner K, Sodeik B, Knebel-Morsdorf D (2018) Entry of herpes simplex virus 1 into epidermis and dermal fibroblasts is independent of the scavenger receptor MARCO. J Virol 92(15):e00490–e00418. https://doi.org/10.1128/JVI.00490-18
Campadelli-Fiume G, Cocchi F, Menotti L, Lopez M (2000) The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev Med Virol 10(5):305–319. https://doi.org/10.1002/1099-1654(200009/10)10:5<305::AID-RMV286>3.0.CO;2-T
Nicoll MP, Proenca JT, Efstathiou S (2012) The molecular basis of herpes simplex virus latency. FEMS Microbiol Rev 36(3):684–705. https://doi.org/10.1111/j.1574-6976.2011.00320.x
Su C, Zhan G, Zheng C (2016) Evasion of host antiviral innate immunity by HSV-1, an update. Virol J 13:38. https://doi.org/10.1186/s12985-016-0495-5
Hsu DY, Shinkai K, Silverberg JI (2018) Epidemiology of eczema herpeticum in hospitalized U.S. children: analysis of a nationwide cohort. J Invest Dermatol 138(2):265–272. https://doi.org/10.1016/j.jid.2017.08.039
Beck LA, Boguniewicz M, Hata T, Schneider LC, Hanifin J, Gallo R, Paller AS, Lieff S, Reese J, Zaccaro D, Milgrom H, Barnes KC, Leung DYM (2009) Phenotype of atopic dermatitis subjects with a history of eczema herpeticum. J Allergy Clin Immunol 124(2):260–269.e267. https://doi.org/10.1016/j.jaci.2009.05.020
Wollenberg A, Zoch C, Wetzel S, Plewig G, Przybilla B (2003) Predisposing factors and clinical features of eczema herpeticum: a retrospective analysis of 100 cases. J Am Acad Dermatol 49(2):198–205 Doi:S019096220300896X
Leung DYM (2013) Why is eczema herpeticum unexpectedly rare? Antivir Res 98(2):153–157. https://doi.org/10.1016/j.antiviral.2013.02.010
Kim K, Kang J, Won Kim S, Sung M (2016) Relationship between the presence of eczema herpeticum and the significance of clinical and laboratory tests in Korean children with atopic dermatitis. Iran J Pediatr 26(4):e4683. https://doi.org/10.5812/ijp.4683
Higaki S, Inoue Y, Yoshida A, Maeda N, Watanabe H, Shimomura Y (2008) Case of bilateral multiple herpetic epithelial keratitis manifested as dendriform epithelial edema during primary Kaposi's varicelliform eruption. Jpn J Ophthalmol 52(2):127–129. https://doi.org/10.1007/s10384-007-0514-6
Ewing CI, Roper HP, David TJ, Haeney MR (1989) Death from eczema herpeticum in a child with severe eczema, mental retardation and cataracts. J R Soc Med 82(3):169–170. https://doi.org/10.1177/014107688908200319
DiCarlo A, Amon E, Gardner M, Barr S, Ott K (2008) Eczema herpeticum in pregnancy and neonatal herpes infection. Obstet Gynecol 112(2 Pt 2):455–457. https://doi.org/10.1097/AOG.0b013e318169ce19
Blanter M, Vickers J, Russo M, Safai B (2015) Eczema Herpeticum: would you know it if you saw it? Pediatr Emerg Care 31(8):586–588. https://doi.org/10.1097/PEC.0000000000000516
Ferrari B, Taliercio V, Luna P, Abad ME, Larralde M (2015) Kaposi's varicelliform eruption: a case series. Indian Dermatol Online J 6(6):399–402. https://doi.org/10.4103/2229-5178.169714
Harindra V, Paffett MC (2001) Recurrent eczema herpeticum: an underrecognised condition. Sex Transm Infect 77(1):76. https://doi.org/10.1136/sti.77.1.76
Zhuang K, Wu Q, Ran X, Ran Y, Ding L, Xu X, Lei S, Lama J (2016) Oral treatment with valacyclovir for HSV-2-associated eczema herpeticum in a 9-month-old infant: a case report. Medicine (Baltimore) 95(29):e4284. https://doi.org/10.1097/MD.0000000000004284
Micali G, Lacarrubba F (2017) Eczema herpeticum. N Engl J Med 377(7):e9. https://doi.org/10.1056/NEJMicm1701668
Jamil A, Muthupalaniappen L (2014) Vesicles and erosions in a patient with chronic eczema: is it just eczema? Ann Acad Med Singap 43(2):130–131
Kim EL, Hohmuth B (2017) Eczema herpeticum in early pregnancy. CMAJ 189(13):E505. https://doi.org/10.1503/cmaj.151544
Shenoy R, Mostow E, Cain G (2015) Eczema herpeticum in a wrestler. Clin J Sport Med 25(1):e18–e19. https://doi.org/10.1097/JSM.0000000000000097
Sahoo B, Handa S, Kumar B (2001) Eczema herpeticum in parthenium dermatitis. Contact Dermatitis 44(2):106–107
Cooper BL (2017) Eczema Herpeticum. J Emerg Med 53(3):412–413. https://doi.org/10.1016/j.jemermed.2016.12.004
Gurvits GE, Nord JA (2011) Eczema herpeticum in pregnancy. Dermatol Reports 3(2):e32. https://doi.org/10.4081/dr.2011.e32
Boyd DA, Sperling LC, Norton SA (2009) Eczema herpeticum and clinical criteria for investigating smallpox. Emerg Infect Dis 15(7):1102–1104. https://doi.org/10.3201/eid1507.090093
Beverido LG, Nanjappa S, Braswell DS, Messina JL, Greene JN (2017) Eczema Herpeticum: a case report and review of literature. Infect Dis Clin Pract 25(2):94–96. https://doi.org/10.1097/ipc.0000000000000471
Celtik C, Karal Y, Kibris A, Kircuval D, Tuzun B (2011) A life-threatening condition in a child with chicken pox: eczema herpeticum. Open J Pediatr 01(01):1–3. https://doi.org/10.4236/ojped.2011.11001
Popov Y, Nikolov R, Lalova A (2010) Localized eczema herpeticum with unilateral ocular involvement. Acta Dermatovenerol Alp Pannonica Adriat 19(3):35–37
Terezhalmy GT, Tyler MT, Ross GR (1979) Eczema herpeticum: atopic dermatitis complicated by primary herpetic gingivostomatitis. Oral Surg Oral Med Oral Pathol 48(6):513–516
Sanderson IR, Brueton LA, Savage MO, Harper JI (1987) Eczema herpeticum: a potentially fatal disease. Br Med J (Clin Res Ed) 294(6573):693–694. https://doi.org/10.1136/bmj.294.6573.693
Finlow C, Thomas J (2018) Disseminated herpes simplex virus: a case of eczema herpeticum causing viral encephalitis. J R Coll Physicians Edinb 48(1):36–39. https://doi.org/10.4997/JRCPE.2018.108
Okamoto M, Takahagi S, Tanaka A, Ogawa A, Nobuki H, Hide M (2018) A case of Kaposi varicelliform eruption progressing to herpes simplex virus hepatitis in an immunocompetent patient. Clin Exp Dermatol 43(5):636–638. https://doi.org/10.1111/ced.13405
Frisch S, Siegfried EC (2011) The clinical spectrum and therapeutic challenge of eczema herpeticum. Pediatr Dermatol 28(1):46–52. https://doi.org/10.1111/j.1525-1470.2010.01356.x
Tupe CL, Weiler BA, Verceles AC, McCurdy MT (2016) A fatal case of eczema herpeticum with septic shock due to methicillin-resistant Staphylococcus aureus. Am J Crit Care 25(4):379–382. https://doi.org/10.4037/ajcc2016495
Karray M, Souissi A (2018) Kaposi Varicelliform eruption. StatPearls [internet]. Doi:NBK482432
Khan MS, Shaw L, Clark V, Afzal Z (2005) Eczema herpeticum: a case report. Int J Paediatr Dent 15(2):136–139. https://doi.org/10.1111/j.1365-263X.2005.00581.x
Ozcan A, Senol M, Saglam H, Seyhan M, Durmaz R, Aktas E, Ozerol IH (2007) Comparison of the Tzanck test and polymerase chain reaction in the diagnosis of cutaneous herpes simplex and varicella zoster virus infections. Int J Dermatol 46(11):1177–1179. https://doi.org/10.1111/j.1365-4632.2007.03337.x
Studdiford JS, Valko GP, Belin LJ, Stonehouse AR (2011) Eczema herpeticum: making the diagnosis in the emergency department. J Emerg Med 40(2):167–169. https://doi.org/10.1016/j.jemermed.2007.11.049
Wheeler CE Jr, Abele DC (1966) Eczema herpeticum, primary and recurrent. Arch Dermatol 93(2):162–173
Piret J, Boivin G (2011) Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob Agents Chemother 55(2):459–472. https://doi.org/10.1128/AAC.00615-10
Kukhanova MK, Korovina AN, Kochetkov SN (2014) Human herpes simplex virus: life cycle and development of inhibitors. Biochemistry (Mosc) 79(13):1635–1652. https://doi.org/10.1134/S0006297914130124
Darji K, Frisch S, Adjei Boakye E, Siegfried E (2017) Characterization of children with recurrent eczema herpeticum and response to treatment with interferon-gamma. Pediatr Dermatol 34(6):686–689. https://doi.org/10.1111/pde.13301
Piret J, Boivin G (2014) Antiviral drug resistance in herpesviruses other than cytomegalovirus. Rev Med Virol 24(3):186–218. https://doi.org/10.1002/rmv.1787
Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, Debenedetto A, Schneider L, Beck LA, Barnes KC, Leung DY (2007) Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol 120(1):150–155. https://doi.org/10.1016/j.jaci.2007.04.031
Thyssen JP, Kezic S (2014) Causes of epidermal filaggrin reduction and their role in the pathogenesis of atopic dermatitis. J Allergy Clin Immunol 134(4):792–799. https://doi.org/10.1016/j.jaci.2014.06.014
O'Regan GM, Sandilands A, McLean WH, Irvine AD (2008) Filaggrin in atopic dermatitis. J Allergy Clin Immunol 122(4):689–693. https://doi.org/10.1016/j.jaci.2008.08.002
Gao P-S, Rafaels NM, Hand T, Murray T, Boguniewicz M, Hata T, Schneider L, Hanifin JM, Gallo RL, Gao L, Beaty TH, Beck LA, Barnes KC, Leung DYM (2009) Filaggrin mutations that confer risk of atopic dermatitis confer greater risk for eczema herpeticum. J Allergy Clin Immunol 124(3):507–513.e507. https://doi.org/10.1016/j.jaci.2009.07.034
De Benedetto A, Slifka MK, Rafaels NM, Kuo IH, Georas SN, Boguniewicz M, Hata T, Schneider LC, Hanifin JM, Gallo RL, Johnson DC, Barnes KC, Leung DYM, Beck LA (2011) Reductions in claudin-1 may enhance susceptibility to herpes simplex virus 1 infections in atopic dermatitis. J Allergy Clin Immunol 128(1):242–246.e245. https://doi.org/10.1016/j.jaci.2011.02.014
De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, Cheadle C, Berger AE, Zhang K, Vidyasagar S, Yoshida T, Boguniewicz M, Hata T, Schneider LC, Hanifin JM, Gallo RL, Novak N, Weidinger S, Beaty TH, Leung DY, Barnes KC, Beck LA (2011) Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol 127(3):773–786 e771-777. https://doi.org/10.1016/j.jaci.2010.10.018
Howell MD, Gao P, Kim BE, Lesley LJ, Streib JE, Taylor PA, Zaccaro DJ, Boguniewicz M, Beck LA, Hanifin JM, Schneider LC, Hata TR, Gallo RL, Kaplan MH, Barnes KC, Leung DYM (2011) The signal transducer and activator of transcription 6 gene (STAT6) increases the propensity of patients with atopic dermatitis toward disseminated viral skin infections. J Allergy Clin Immunol 128(5):1006–1014. https://doi.org/10.1016/j.jaci.2011.06.003
Gao P-S, Rafaels NM, Mu D, Hand T, Murray T, Boguniewicz M, Hata T, Schneider L, Hanifin JM, Gallo RL, Gao L, Beaty TH, Beck LA, Leung DYM, Barnes KC (2010) Genetic variants in thymic stromal lymphopoietin are associated with atopic dermatitis and eczema herpeticum. J Allergy Clin Immunol 125(6):1403–1407.e1404. https://doi.org/10.1016/j.jaci.2010.03.016
Gao P-S, Leung DYM, Rafaels NM, Boguniewicz M, Hand T, Gao L, Hata TR, Schneider LC, Hanifin JM, Beaty TH, Beck LA, Weinberg A, Barnes KC (2012) Genetic variants in interferon regulatory factor 2 (IRF2) are associated with atopic dermatitis and eczema herpeticum. J Invest Dermatol 132(3):650–657. https://doi.org/10.1038/jid.2011.374
Leung DYM, Gao P-S, Grigoryev DN, Rafaels NM, Streib JE, Howell MD, Taylor PA, Boguniewicz M, Canniff J, Armstrong B, Zaccaro DJ, Schneider LC, Hata TR, Hanifin JM, Beck LA, Weinberg A, Barnes KC (2011) Human atopic dermatitis complicated by eczema herpeticum is associated with abnormalities in IFN-γ response. J Allergy Clin Immunol 127(4):965–973.e965. https://doi.org/10.1016/j.jaci.2011.02.010
Kim BE, Bin L, Ye Y-M, Ramamoorthy P, Leung DYM (2013) IL-25 enhances HSV-1 replication by inhibiting filaggrin expression, and acts synergistically with Th2 cytokines to enhance HSV-1 replication. J Invest Dermatol 133(12):2678–2685. https://doi.org/10.1038/jid.2013.223
Rosenthal KS, Killius J, Hodnichak CM, Venetta TM, Gyurgyik L, Janiga K (1989) Mild acidic pH inhibition of the major pathway of herpes simplex virus entry into HEp-2 cells. J Gen Virol 70(Pt 4):857–867. https://doi.org/10.1099/0022-1317-70-4-857
Niessen CM (2007) Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol 127(11):2525–2532. https://doi.org/10.1038/sj.jid.5700865
Rahn E, Thier K, Petermann P, Rubsam M, Staeheli P, Iden S, Niessen CM, Knebel-Morsdorf D (2017) Epithelial barriers in murine skin during herpes simplex virus 1 infection: the role of tight junction formation. J Invest Dermatol 137(4):884–893. https://doi.org/10.1016/j.jid.2016.11.027
Yoon M, Spear PG (2002) Disruption of adherens junctions liberates nectin-1 to serve as receptor for herpes simplex virus and pseudorabies virus entry. J Virol 76(14):7203–7208. https://doi.org/10.1128/jvi.76.14.7203-7208.2002
Bardan A, Nizet V, Gallo RL (2004) Antimicrobial peptides and the skin. Expert Opin Biol Ther 4(4):543–549. https://doi.org/10.1517/14712598.4.4.543
Harder J, Dressel S, Wittersheim M, Cordes J, Meyer-Hoffert U, Mrowietz U, Folster-Holst R, Proksch E, Schroder JM, Schwarz T, Glaser R (2010) Enhanced expression and secretion of antimicrobial peptides in atopic dermatitis and after superficial skin injury. J Invest Dermatol 130(5):1355–1364. https://doi.org/10.1038/jid.2009.432
Roy M, Lebeau L, Chessa C, Damour A, Ladram A, Oury B, Boutolleau D, Bodet C, Leveque N (2019) Comparison of anti-viral activity of frog skin anti-microbial peptides Temporin-Sha and [K(3)]SHa to LL-37 and Temporin-Tb against herpes simplex virus type 1. Viruses 11(1). https://doi.org/10.3390/v11010077
Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DY (2002) Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med 347(15):1151–1160. https://doi.org/10.1056/NEJMoa021481
Howell MD, Boguniewicz M, Pastore S, Novak N, Bieber T, Girolomoni G, Leung DY (2006) Mechanism of HBD-3 deficiency in atopic dermatitis. Clin Immunol 121(3):332–338. https://doi.org/10.1016/j.clim.2006.08.008
Howell M, Wollenberg A, Gallo R, Flaig M, Streib J, Wong C, Pavicic T, Boguniewicz M, Leung D (2006) Cathelicidin deficiency predisposes to eczema herpeticum. J Allergy Clin Immunol 117(4):836–841. https://doi.org/10.1016/j.jaci.2005.12.1345
Hata TR, Kotol P, Boguniewicz M, Taylor P, Paik A, Jackson M, Nguyen M, Kabigting F, Miller J, Gerber M, Zaccaro D, Armstrong B, Dorschner R, Leung DY, Gallo RL (2010) History of eczema herpeticum is associated with the inability to induce human beta-defensin (HBD)-2, HBD-3 and cathelicidin in the skin of patients with atopic dermatitis. Br J Dermatol 163(3):659–661. https://doi.org/10.1111/j.1365-2133.2010.09892.x
Wilson SS, Wiens ME, Smith JG (2013) Antiviral mechanisms of human defensins. J Mol Biol 425(24):4965–4980. https://doi.org/10.1016/j.jmb.2013.09.038
Hazrati E, Galen B, Lu W, Wang W, Ouyang Y, Keller MJ, Lehrer RI, Herold BC (2006) Human alpha- and beta-defensins block multiple steps in herpes simplex virus infection. J Immunol 177(12):8658–8666 Doi:177/12/8658
Muraro A, Lemanske RF Jr, Hellings PW, Akdis CA, Bieber T, Casale TB, Jutel M, Ong PY, Poulsen LK, Schmid-Grendelmeier P, Simon HU, Seys SF, Agache I (2016) Precision medicine in patients with allergic diseases: airway diseases and atopic dermatitis-PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol 137(5):1347–1358. https://doi.org/10.1016/j.jaci.2016.03.010
Gittler JK, Shemer A, Suarez-Farinas M, Fuentes-Duculan J, Gulewicz KJ, Wang CQ, Mitsui H, Cardinale I, de Guzman SC, Krueger JG, Guttman-Yassky E (2012) Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol 130(6):1344–1354. https://doi.org/10.1016/j.jaci.2012.07.012
Guttman-Yassky E, Krueger JG (2017) Atopic dermatitis and psoriasis: two different immune diseases or one spectrum? Curr Opin Immunol 48:68–73. https://doi.org/10.1016/j.coi.2017.08.008
Tsakok T, Woolf R, Smith CH, Weidinger S, Flohr C (2018) Atopic dermatitis: the skin barrier and beyond. Br J Dermatol 180(3):464–474. https://doi.org/10.1111/bjd.16934
Brandt EB, Sivaprasad U (2011) Th2 cytokines and atopic dermatitis. J Clin Cell Immunol 2(3):110. https://doi.org/10.4172/2155-9899.1000110
Wollenberg A, Räwer H-C, Schauber J (2010) Innate immunity in atopic dermatitis. Clin Rev Allergy Immunol 41(3):272–281. https://doi.org/10.1007/s12016-010-8227-x
Traidl S, Kienlin P, Begemann G, Jing L, Koelle DM, Werfel T, Roesner LM (2018) Patients with atopic dermatitis and history of eczema herpeticum elicit herpes simplex virus-specific type 2 immune responses. J Allergy Clin Immunol 141(3):1144–1147 e1145. https://doi.org/10.1016/j.jaci.2017.09.048
Hvid M, Vestergaard C, Kemp K, Christensen GB, Deleuran B, Deleuran M (2011) IL-25 in atopic dermatitis: a possible link between inflammation and skin barrier dysfunction? J Invest Dermatol 131(1):150–157. https://doi.org/10.1038/jid.2010.277
Tatsuno K, Fujiyama T, Yamaguchi H, Waki M, Tokura Y (2015) TSLP directly interacts with skin-homing Th2 cells highly expressing its receptor to enhance IL-4 production in atopic dermatitis. J Invest Dermatol 135(12):3017–3024. https://doi.org/10.1038/jid.2015.318
Oyoshi MK, Venturelli N, Geha RS (2016) Thymic stromal lymphopoietin and IL-33 promote skin inflammation and vaccinia virus replication in a mouse model of atopic dermatitis. J Allergy Clin Immunol 138(1):283–286. https://doi.org/10.1016/j.jaci.2015.12.1304
Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K (2005) Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med 202(5):637–650. https://doi.org/10.1084/jem.20050821
Bogdan C (2000) The function of type I interferons in antimicrobial immunity. Curr Opin Immunol 12(4):419–424 Doi:S0952-7915(00)00111-4
Schoggins JW, Rice CM (2011) Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 1(6):519–525. https://doi.org/10.1016/j.coviro.2011.10.008
Mikloska Z, Cunningham AL (2001) Alpha and gamma interferons inhibit herpes simplex virus type 1 infection and spread in epidermal cells after axonal transmission. J Virol 75(23):11821–11826. https://doi.org/10.1128/JVI.75.23.11821-11826.2001
Bin L, Edwards MG, Heiser R, Streib JE, Richers B, Hall CF, Leung DYM (2014) Identification of novel gene signatures in patients with atopic dermatitis complicated by eczema herpeticum. J Allergy Clin Immunol 134(4):848–855. https://doi.org/10.1016/j.jaci.2014.07.018
Bin L, Li X, Richers B, Streib JE, Hu JW, Taylor P, Leung DYM (2018) Ankyrin repeat domain 1 regulates innate immune responses against herpes simplex virus 1: a potential role in eczema herpeticum. J Allergy Clin Immunol 141(6):2085–2093 e2081. https://doi.org/10.1016/j.jaci.2018.01.001
Wollenberg A, Wagner M, Gunther S, Towarowski A, Tuma E, Moderer M, Rothenfusser S, Wetzel S, Endres S, Hartmann G (2002) Plasmacytoid dendritic cells: a new cutaneous dendritic cell subset with distinct role in inflammatory skin diseases. J Invest Dermatol 119(5):1096–1102. https://doi.org/10.1046/j.1523-1747.2002.19515.x
Frey KG, Ahmed CM, Dabelic R, Jager LD, Noon-Song EN, Haider SM, Johnson HM, Bigley NJ (2009) HSV-1-induced SOCS-1 expression in keratinocytes: use of a SOCS-1 antagonist to block a novel mechanism of viral immune evasion. J Immunol 183(2):1253–1262. https://doi.org/10.4049/jimmunol.0900570
Wolk K, Witte K, Witte E, Raftery M, Kokolakis G, Philipp S, Schonrich G, Warszawska K, Kirsch S, Prosch S, Sterry W, Volk HD, Sabat R (2013) IL-29 is produced by T(H)17 cells and mediates the cutaneous antiviral competence in psoriasis. Sci Transl Med 5(204):204ra129. https://doi.org/10.1126/scitranslmed.3006245
Staudacher A, Hinz T, Novak N, von Bubnoff D, Bieber T (2015) Exaggerated IDO1 expression and activity in Langerhans cells from patients with atopic dermatitis upon viral stimulation: a potential predictive biomarker for high risk of eczema herpeticum. Allergy 70(11):1432–1439. https://doi.org/10.1111/all.12699
Adams O, Besken K, Oberdorfer C, MacKenzie CR, Russing D, Daubener W (2004) Inhibition of human herpes simplex virus type 2 by interferon gamma and tumor necrosis factor alpha is mediated by indoleamine 2,3-dioxygenase. Microbes Infect 6(9):806–812. https://doi.org/10.1016/j.micinf.2004.04.007
Topham NJ, Hewitt EW (2009) Natural killer cell cytotoxicity: how do they pull the trigger? Immunology 128(1):7–15. https://doi.org/10.1111/j.1365-2567.2009.03123.x
Nandakumar S, Woolard SN, Yuan D, Rouse BT, Kumaraguru U (2008) Natural killer cells as novel helpers in anti-herpes simplex virus immune response. J Virol 82(21):10820–10831. https://doi.org/10.1128/JVI.00365-08
Goodyear HM, McLeish P, Randall S, Buchan A, Skinner GR, Winther M, Rolland J, Morgan G, Harper JI (1996) Immunological studies of herpes simplex virus infection in children with atopic eczema. Br J Dermatol 134(1):85–93
Kawakami Y, Ando T, Lee J-R, Kim G, Kawakami Y, Nakasaki T, Nakasaki M, Matsumoto K, Choi YS, Kawakami T (2017) Defective natural killer cell activity in a mouse model of eczema herpeticum. J Allergy Clin Immunol 139(3):997–1006.e1010. https://doi.org/10.1016/j.jaci.2016.06.034
Fernandez MA, Puttur FK, Wang YM, Howden W, Alexander SI, Jones CA (2008) T regulatory cells contribute to the attenuated primary CD8+ and CD4+ T cell responses to herpes simplex virus type 2 in neonatal mice. J Immunol 180(3):1556–1564 Doi:180/3/1556
Takahashi R, Sato Y, Kurata M, Yamazaki Y, Kimishima M, Shiohara T (2013) Pathological role of regulatory T cells in the initiation and maintenance of eczema herpeticum lesions. J Immunol 192(3):969–978. https://doi.org/10.4049/jimmunol.1300102
Lyons JJ, Milner JD, Stone KD (2015) Atopic dermatitis in children: clinical features, pathophysiology, and treatment. Immunol Allergy Clin N Am 35(1):161–183. https://doi.org/10.1016/j.iac.2014.09.008
Lubbe J, Pournaras CC, Saurat JH (2000) Eczema herpeticum during treatment of atopic dermatitis with 0.1% tacrolimus ointment. Dermatology 201(3):249–251. https://doi.org/10.1159/000018497
Segura S, Romero D, Carrera C, Iranzo P, Estrach T (2005) Eczema herpeticum during treatment of atopic dermatitis with 1% pimecrolimus cream. Acta Derm Venereol 85(6):524–525. https://doi.org/10.1080/00015550510034164
Wahn U, Bos JD, Goodfield M, Caputo R, Papp K, Manjra A, Dobozy A, Paul C, Molloy S, Hultsch T, Graeber M, Cherill R, de Prost Y (2002) Efficacy and safety of pimecrolimus cream in the long-term management of atopic dermatitis in children. Pediatrics 110(1 Pt 1):e2. https://doi.org/10.1542/peds.110.1.e2
Reitamo S, Wollenberg A, Schopf E, Perrot JL, Marks R, Ruzicka T, Christophers E, Kapp A, Lahfa M, Rubins A, Jablonska S, Rustin M (2000) Safety and efficacy of 1 year of tacrolimus ointment monotherapy in adults with atopic dermatitis. The European Tacrolimus Ointment Study Group. Arch Dermatol 136(8):999–1006 doi:dst0016
Koo JY, Fleischer AB Jr, Abramovits W, Pariser DM, McCall CO, Horn TD, Gottlieb AB, Jaracz E, Rico MJ (2005) Tacrolimus ointment is safe and effective in the treatment of atopic dermatitis: results in 8000 patients. J Am Acad Dermatol 53(2 Suppl 2):S195–S205. https://doi.org/10.1016/j.jaad.2005.04.063
Reitamo S, Rustin M, Harper J, Kalimo K, Rubins A, Cambazard F, Brenninkmeijer EE, Smith C, Berth-Jones J, Ruzicka T, Sharpe G, Taieb A (2008) A 4-year follow-up study of atopic dermatitis therapy with 0.1% tacrolimus ointment in children and adult patients. Br J Dermatol 159(4):942–951. https://doi.org/10.1111/j.1365-2133.2008.08747.x
Kim SW, Park YW, Kwon IH, Kim KH (2010) Cyclosporin treatment of atopic dermatitis: is it really associated with infectious diseases? Ann Dermatol 22(2):170–172. https://doi.org/10.5021/ad.2010.22.2.170
Fleming P, Drucker AM (2018) Risk of infection in patients with atopic dermatitis treated with Dupilumab: a meta-analysis of randomized controlled trials. J Am Acad Dermatol 78(1):62–69 e61. https://doi.org/10.1016/j.jaad.2017.09.052
Grice EA, Segre JA (2011) The skin microbiome. Nat Rev Microbiol 9(4):244–253. https://doi.org/10.1038/nrmicro2537
Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Bouffard GG, Blakesley RW, Murray PR, Green ED, Turner ML, Segre JA (2009) Topographical and temporal diversity of the human skin microbiome. Science 324(5931):1190–1192. https://doi.org/10.1126/science.1171700
Scharschmidt TC, Fischbach MA (2013) What lives on our skin: ecology, genomics and therapeutic opportunities of the skin microbiome. Drug Discov Today Dis Mech 10(3–4):e83–e89. https://doi.org/10.1016/j.ddmec.2012.12.003
Yamazaki Y, Nakamura Y, Nunez G (2017) Role of the microbiota in skin immunity and atopic dermatitis. Allergol Int 66(4):539–544. https://doi.org/10.1016/j.alit.2017.08.004
Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, Nomicos E, Polley EC, Komarow HD, Murray PR, Turner ML, Segre JA (2012) Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res 22(5):850–859. https://doi.org/10.1101/gr.131029.111
Bjerre RD, Bandier J, Skov L, Engstrand L, Johansen JD (2017) The role of the skin microbiome in atopic dermatitis: a systematic review. Br J Dermatol 177(5):1272–1278. https://doi.org/10.1111/bjd.15390
Oh JE, Kim BC, Chang DH, Kwon M, Lee SY, Kang D, Kim JY, Hwang I, Yu JW, Nakae S, Lee HK (2016) Dysbiosis-induced IL-33 contributes to impaired antiviral immunity in the genital mucosa. Proc Natl Acad Sci U S A 113(6):E762–E771. https://doi.org/10.1073/pnas.1518589113
Bin L, Kim BE, Brauweiler A, Goleva E, Streib J, Ji Y, Schlievert PM, Leung DY (2012) Staphylococcus aureus alpha-toxin modulates skin host response to viral infection. J Allergy Clin Immunol 130(3):683–691 e682. https://doi.org/10.1016/j.jaci.2012.06.019
Hepburn L, Hijnen DJ, Sellman BR, Mustelin T, Sleeman MA, May RD, Strickland I (2017) The complex biology and contribution of Staphylococcus aureus in atopic dermatitis, current and future therapies. Br J Dermatol 177(1):63–71. https://doi.org/10.1111/bjd.15139
Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Munoz-Planillo R, Hasegawa M, Villaruz AE, Cheung GY, McGavin MJ, Travers JB, Otto M, Inohara N, Nunez G (2013) Staphylococcus delta-toxin induces allergic skin disease by activating mast cells. Nature 503(7476):397–401. https://doi.org/10.1038/nature12655
Syed AK, Reed TJ, Clark KL, Boles BR, Kahlenberg JM (2015) Staphlyococcus aureus phenol-soluble modulins stimulate the release of proinflammatory cytokines from keratinocytes and are required for induction of skin inflammation. Infect Immun 83(9):3428–3437. https://doi.org/10.1128/IAI.00401-15
Acknowledgments
We thank Jeffrey Arsham for the English revision of the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This manuscript complies with the ethical standards of Clinical Reviews in Allergy and Immunology.
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
No ethics issues are raised as this is a review article.
Informed Consent
Informed consent was obtained from all patients for clinical photographs included in this review article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Damour, A., Garcia, M., Seneschal, J. et al. Eczema Herpeticum: Clinical and Pathophysiological Aspects. Clinic Rev Allerg Immunol 59, 1–18 (2020). https://doi.org/10.1007/s12016-019-08768-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-019-08768-3