
Abstract
The aim of this article is to present an overview of impurities of alkali metals and alkaline earth elements included in smelter-grade alumina and other raw materials that primarily report to the electrolyte, i.e., bath, and to suggest pathways for improved control at primary aluminum smelters. Comments are included on how these impurities affect intermediate subprocesses, metal products, and cell condition. The authors have worked to create a reference that not only reports on the scientific aspects of how impurities react in a bath of molten salt under electrolysis but also covers the practical matters of concern regarding impurities that affect the electrolyte. By combining scientific knowledge with process engineering techniques, readers should be able to better manage impurities and related impacts in aluminum production processes.
Similar content being viewed by others

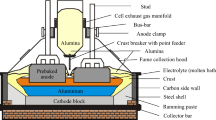
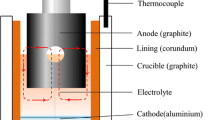
Introduction
Impurities have importance. This is evident in the pricing of various grades of as-cast Al ingot traded by the London Metal Exchange (LME) on through to the wide array of aluminum products that demand very high-purity (99.9x% or greater) Al or various alloys that contain tightly controlled additions of, e.g., magnesium, silicon, copper, or zinc for the production of wrought alloys.
The published literature on the role of impurities in the operating cells has often focused on one specific element, such as phosphorus1 and its impact on energy efficiency, which is only one metric in operating a successful and profitable smelter. This is often not the case with many other impurities, especially when attempting to fine-tune cell operations for performance. As is well known, the extraction of aluminum from its oxide is efficiently performed electrochemically with the use of a consumable carbon anode, and an electrochemically stable solvent, molten salt, electrolyte that dissolves smelter-grade alumina (SGA) produced by refineries. Subject to constraints of the electrochemical process operating condition, the dominant electrochemical reactions produce liquid aluminum at the cathode and a mix of gaseous products released from the anode dominated by carbon dioxide, carbonyl sulfide, and carbon monoxide.
But what happens to impurities when they enter the electrolyte? The old adage “What goes in, must come out!” also applies to the Hall–Héroult process.
There are four sources of “impurities,” the terminology assigned to constituents of the raw materials entering the cell other than pure Al2O3 and pure carbon. These sources are:
-
a.
Other oxides that are entrapped or coextracted during production of SGA.
-
b.
Elements present in hydrocarbon structures that have been thermally decomposed into calcined petroleum coke providing the carbon source for anodes. Assembled anodes contain additional contaminants picked up or entrapped during production processes.
-
c.
Corrosion products and dissolution products arising from the cell lining and construction components that are attacked by the electrolyte or the metal, either chemically or electrochemically.
-
d.
Other contaminants not directly related to raw materials.
The third category, (c), often includes corrosion products from chemical or electrochemical oxidation of anode stubs when they make direct electrical contact with the electrolyte. Then, electrochemically oxidized products find their way into the electrolyte. Other impurities linked to anodes are formed from slow chemical interactions occurring with hot anode cover material (ACM), and these can enter the electrolyte through spillage of subsequently recycled ACM.
Impurities of the fourth category, (d), enter by various paths and may include concrete and brick pieces from floors and pot line basements, material recycled from the cast house to the smelter, and recovery of material skimmed from pots. This category may even include dust that settles out of ambient air. Unfortunately, then the operating cell can become a convenient “dustbin.”
All of these materials eventually make contact with the electrolyte, and this has a strong influence on what happens to the impurity, either immediately or over time. Because of the stability of alumina, and the design restrictions associated with the need to use consumable carbon anodes, there is no perfect electrolyte that fulfills all of the ideal requirements. The “wish list” includes:
-
The electrolyte must dissolve the reactant, normally fluorinated SGA, sufficiently to minimize concentration polarization enabling adequate current density. This immediately constrains it to molten fluoride salts such as cryolite.
-
The electrolyte must be thermochemically stable in the operating environment so that it does not adversely contaminate the metal product.
-
The electrolyte must be a reasonably good electrical conductor to enable adequate separation between the two electrochemical products such that chemical back-reaction is prevented.
-
The electrolyte should not readily dissolve the electrode products to prevent efficiency losses occurring through chemical backreaction.
-
The electrolyte must have a specific gravity that is low enough for the product to settle beneath it and to remain reasonably separated from it.
-
The electrolyte should have as low a liquidus temperature as is practical to minimize heat loss and thermal energy requirements, opening the door to intentional additions of impurities, such as CaF2, to electrolytic bath.2
No mixture of molten salts fully satisfies all of these requirements. Based on experience and design constraints, a molten mixture of NaF–AlF3–CaF2 satisfies most. The primary weakness is that, in the presence of pure aluminum, there is limited interaction with the electrolyte, represented by the following reaction:
Detailed studies presented by Tingle et al.,3 who equilibrated NaF–AlF3 mixtures with pure aluminum, showed that the equilibrium portion of sodium in aluminum could vary by more than an order of magnitude for changes in the interfacial ratio of NaF/AlF3 and temperature that occur with different cell designs and process operating conditions used in the smelting industry. Figure 1 shows a representation of this variation encompassing the extremes experienced for different smelter designs. While smelters typically operate with molar ratios of between 2.2 and 2.6, because of the interfacial enrichment through cathode-polarization-generated Na+ transport of current, the interfacial ratio is always higher and can exceed 3.0, especially at higher cathode current densities and in well magnetically compensated cells. The oval red band encompasses interfacial measurements resulting in different cell designs.

adapted from Tingle et al.3).
Comparing the variation in minimum (equilibrium) Na content of Al with “cryolite” wt. ratio and temperature (
This means that such an alloy would automatically be produced at the reversible potential of which the aluminum is being formed. However, with the sodium ion (Na+) being the main charge carrier in the electrolyte, the proportion of Na (in Al) codeposited will be significantly higher. Unfortunately, molten alkali fluorides also dissolve metals of their parent salt, which we are familiar with by the accepted mechanism of current efficiency loss.
While Na can be readily removed from the metal product using appropriate treatment methodology, its deposition in Al simultaneously leads to Na being dissolved in the electrolyte according to the following interfacial equilibrium:
Not surprisingly, the work of Tingle et al. has shown that the dissolved metal in the electrolyte is dominantly sodium. This has also been clearly established by the earlier research of Grjotheim4 and that of Snow and Welch.5 Note that another problem arises with the dissolved Na, viz. the creation of a reducing media in the electrolyte for interaction with impurities such as phosphorus pentoxide that are introduced to the cell by various mechanisms.
When developing molten salts as coolants for nuclear reactors, Dworkin et al.6 performed an extensive study of the solubility of alkali metals in their molten fluoride. One of the resulting phase diagrams generated is reproduced in part in Fig. 2. While the values are not directly transferable because the concentration of the alkali fluorides is lower in the aluminum smelting cell, it also points to a disadvantage of using potassium cryolite equivalents.

adapted from Dworkin et al.6).
Phase diagram of the solubility of alkali metals in their molten fluorides (
Based on operating experience in modern smelters and for the efficient use of all resources, including electrical energy, the chemical composition of the solvent electrolyte universally used has only a limited operating band. The electrolyte composition universally targeted results from mixing NaF, AlF3, and CaF2 within the following composition limits:
-
49 wt.% to 52 wt.% sodium fluoride (NaF)
-
44 wt.% to 45 wt.% aluminum fluoride (AlF3)
-
4 wt.% to 6 wt.% calcium fluoride (CaF2)
This is equivalent to a cryolite ratio of between 1.08 and 1.20, and calcium fluoride concentration of 4–6 wt.%, for an operating temperature between 955 °C and 965°C. CaF2 is used to lower the operating temperature because it is extremely stable, with negligible codeposition, but it also tends to dissolve oxides. The upper concentration limit exists to avoid increasing the specific gravity of the electrolyte beyond an acceptable limit.
At times, other compositions have been used that included LiF and MgF2. Additives to produce these are no longer in use at most smelting locations. However, these additives will be discussed briefly in the sections that follow.
Within this predominantly ionic, molten electrolyte, there are Coulombic forces of attraction generating ionic clusters that lead to the formation of other compounds when energy is reduced by cooling below the liquidus point. Because of this, we sometimes refer to the electrolyte composition relative to the species that crystalize from the solution. Based on model predictions from physicochemical measurements, the dominant ionic species in the electrolyte are presented in Table I.
Thermochemically, this means that the electrolyte can be treated as a mixture of CaF2, NaF, AlF3, NaAlF4, Na3AlF6, NaOH, and NaAlOF4 when looking at possible interactions with impurities, to assess what transformations they may undergo.
Smelter-Grade Alumina: Impurity Interactions with Electrolyte
Smelter-grade alumina is produced globally from a variety of different quality bauxites, which often require differing processing conditions. Each bauxite source contains an array of impurities.
The hydroxylated structures of alumina, boehmite AlO(OH) or gibbsite Al(OH)3, are extracted from bauxitic ore bodies by preferential dissolution in concentrated caustic liquors using the Bayer process. Most of the other constituent minerals of the ore bodies have limited solubility. However, depending on the ore body, processing conditions, equipment, and technology of the refinery, limited amounts of other elements such as Na, Ca, K, P, Li, Be, and multiple trace impurities can also find themselves entrapped in the structure of the Al(OH)3 fed to calcination. Additional impurities are deposited on particles of SGA via carryover of contaminants from calcination fuel.7 Most notably these include sulfur, vanadium, and phosphorus. When added to the cell, these impurities dissolve, being transformed to fluorides and oxyfluorides, or they continue to exist as oxides. These can then undergo chemical or electrochemical reactions with the cathodic metal pad, with carbon dust, or as ions in the electrolyte.
Table II presents a summary and composition range of the more dominant impurities found in SGA marketed around the world. Particular focus is placed on impurities that accumulate in the electrolyte.
The impurities that have very stable fluorides chemically interact with the electrolyte and stabilize the cationic species. This is exemplified by CaO in alumina that is converted to CaF2 by the reaction
The substantial reduction in the Gibbs energy (ΔGo960 C) demonstrates that CaF2 is much more stable than CaO in the electrolyte. Other species may undergo an electrochemical reaction with the cation, being codeposited with aluminum or forming a new product with electroactive cell lining materials. Chemical reactions, perhaps with dissolved metal, that transform it into a new product are also possible. The transformed products can leave with exhaust gas, metal, or even by penetrating the cell lining.
None of these options is attractive; environmental issues can arise from gaseous products, metal quality potentially deteriorates from alloying, cell life is reduced by chemical interactions, and all of the electrolyte in the smelter will gradually drift away from specifications through impurity accumulation.
On the basis of a thermodynamic analysis of the properties of the various impurities and electrolyte ingredients, it becomes evident that alkali oxides form stable fluoride products and accumulate in the electrolyte on dissolution, as does the fluoride formed from MgO and CaO (Table III).
However, the conversion of BeO to BeF2 does not have the same driving force when the product is condensed. Beryllium fluoride tends to sublime just above 900°C, and some formation of gaseous products is strongly favored. Consequently, it is not surprising that the highest concentrations of Be compounds in cathodes can be found in the cooler, upper regions of side linings where it has desublimed8 (Fig. 3).

Concentrations of Be in cathode materials (adapted from8).
Other oxides such as SiO2, Fe2O3, and Ga2O3 do not have the same driving force to convert to fluorides. However, Fe2O3 and Ga2O3 are readily reduced by dissolved sodium codepositing Fe and Ga on the Al cathode.
Modern cells typically contain 1.5–1.8 tons of electrolyte for each ton of metal that is produced per day. Each ton of Al produced consumes approximately 1.92 kg of primary smelter grade alumina, in which there are approximately 30 kg that is not Al2O3. Hypothetically, if all the impurities stayed in the electrolyte, its mass would increase by more than 50% in one month. Fortunately it does not, but accumulation is an issue when endeavoring to maintain the electrolyte at its optimum performance composition over many years.
The “impurities” that form thermodynamically stable compounds by interaction may have little or no impact on the metal, but they do affect the process by altering electrolyte composition. The cumulative effect of increasing the concentration of CaF2 in the electrolyte by the reaction given in Eq. (3) will be to lower the liquidus temperature, but also lowering the conductivity of the electrolyte and increasing its density. Operational consequences are:
-
Increasing the superheat, risking loss of protective side ledge, leaving the carbon lining materials vulnerable to degradation and reduced cell life.
-
Higher density risks mixing of electrolyte and metal and even metal pad inversions.
-
Higher electrical resistance automatically reduces anode–cathode distance, lowering current efficiency, increasing operating costs, and changing process economics.9
In summary, impurities do not only affect metal quality. They can also have a broader impact on operations and economics and, in some cases, as exemplified by Eq. (3), are beneficial. There are also electrolyte losses associated with cell design and work practices to consider. As such, the oxide impurities of Na and Ca provide a convenient means to generate “make-up” electrolytes. Each technology has its own idiosyncrasies, but a detailed understanding of the sources and destinies of each impurity is a cornerstone of good operation.
Alkali Metals and Alkaline Earth Elements
Because of their tendency to convert to highly stable fluorides in the molten electrolyte, elements such as sodium, calcium, and potassium have a very high affinity to concentrate in bath. The same applies to lithium and to a lesser extent beryllium and magnesium, although these last three examples generally enter in much smaller quantities.
When alkali metals and alkaline earth elements enter the electrolyte, there are no commercially viable technologies to then separate them out. Accordingly, special attention must be paid in the analysis and approval procedures for raw materials as well as secondary materials that enter the process. Secondary materials may include bath purchases and materials that cast houses may use that then can arrive at the bath treatment plant, including various forms of dross.
Sodium
While sodium is found in calcined petroleum coke and AlF3, the major inputs of Na to reduction cells are from SGA and additions of sodium carbonate, Na2CO3, i.e., “soda ash.” The sodium oxide (Na2O) input in SGA usually exceeds the sum of all other sodium inputs. Adding soda ash eventually creates a financial obligation for adding sufficient AlF3 to neutralize the Na input (Fig. 4). In a few cases, the SGA source may have Na2O levels that are below the point of equilibrium for excess bath generation. Smelters in this situation generally consume bath that has been generated by other smelters.

Sodium oxide in alumina sources versus excess bath generation.
Sodium codeposition into cathodes causes them to swell, crack, and suffer damage that can limit pot life and affect cell performance.10 It also degrades the properties of thermal insulation built into cathodes, negatively affecting energy consumption.
Sodium primarily enters anodes with butts that have not been thoroughly cleaned. Anodes with low levels of sodium have less than 200 ppm Na. Green petroleum coke (GPC) contains 40–120 ppm Na (average of approx. 77 ppm). Sodium concentrations in calcined petroleum coke (CPC) are similar. This places Na in butts as the major contributor under almost any set of circumstances (Fig. 5).

Na in green petroleum coke fractions over and under 1 mm in size.
Sodium penetrates the anodes intercalating Na into its structure. This occurs especially during the brief periods when the anodes come into contact with liquid metal or are not electroactive. It has been established that air reactivity increases with increasing Na concentration in the anodes.11 Furthermore, it has also been observed that Na progressively destroys the refractory linings of anode baking furnaces.
When considering sodium inputs and outputs, the amount of sodium in SGA typically overwhelms the amount of Na that leaves with metal production by about 40:1. It is also well known that sodium intercalates into cathodes, but the bulk of Na2O input stemming from the SGA accumulates in the electrolyte as its fluoride (Table III).
Metal that is siphoned from the reduction cells pots is typically between 50 ppm and 200 ppm Na. Products such as sheet ingots (i.e., rolling slabs) require such concentrations to be reduced further by treatment prior to casting so that the metal has acceptable physical properties. Elevated levels of Na in primary metal also pose a risk to the refractory linings of holding furnaces by chemical attack.
Sodium concentrations over 6 ppm in metal can lead to billet surface defects in 6xxx series products. At levels over 2.0 ppm, it can cause 5xxx series slab to suffer edge cracking issues during rolling. A number of treatment processes may be employed to remove alloyed sodium from aluminum metal. These include Treatment of Aluminum in a Crucible (TAC), charcoal filtration, fluxing with chlorine gas, and the use of various salts. Then, degassing of the metal may need to occur prior to casting.12 However, the concentration of Na in common, LME-grade ingot is not clearly specified, unlike Si and Fe content. Sodium contamination falls into the others each and others total categories, as do many other trace impurities.
At the carbonaceous cathode interface, a significant parallel reaction occurs during the start-up and operation of new cells, i.e., codeposition of Al and Na. The sodium deposition forms an intercalation compound with the carbon according to the overall reaction:
While a film of electrolyte exists between the metal and the carbon surface, with the latter not being saturated in NaCx, its formation continues by an increasing sodium codeposition driving force through the strongly depolarizing effect of the sodium being electrochemically oxidized (but not precluding aluminum also being electrochemically oxidized) anodically
at the metal undersurface,
and no applied voltage is required for this overall reaction. It is essentially its own battery.
Accurate thermochemical data do not exist for calculating the potential, but it is probably below that for aluminum deposition. This is clearly indicated by the rate of change in the cryolite ratio during the first few hours when starting a new cell. Na (in the cathode) typically accounts for more than one-quarter of the electrochemical cathodic reduction products made during the first day of operation based on the amount of Na2CO3 that is added and changes in the cryolite ratio of the electrolyte.
The intercalation rate slows as the interfacial surface becomes concentrated. But it still continues, with the sodium moving down further into the cathode lining via a vapor transport mechanism.13 It does not reach steady state for a number of months. Thereafter, it continues as Na vapor condenses in the cooler regions of the insulation and often under the relatively cool collector bars.
We think of frozen side ledge as a means to prevent attack upon refractory materials used in the side walls of cells. The thickness of side ledge plus the quality of sidewall insulation is determined by the heat loss through the sides of the cell. But the driving force for heat loss is the superheat of the cell, which changes with operating voltage, metal production rate (current efficiency), or electrolyte composition at fixed line current. Only a few degrees of change in superheat can profoundly affect the amount of side ledge. But the most susceptible part for operation without any side ledge is the metal pad zone. When the ledge is lost in this zone, it opens the door to a strong driving force for three processes that negatively impact cell and metal conditions:
-
1.
The first is to enable direct or indirect deposition of sodium into the carbon component of the cell lining. With a large temperature gradient, the sodium moves rapidly through the side wall by the mechanism already discussed. In Fig. 6a, the jet-black area shows NaOH formed by moisture uptake from the air during the cell de-lining process. The second figure, Fig. 6b, is a burning ball of sodium metal that had previously condensed in a cold zone under a collector bar, but self-ignited after having its protective oxide layer disturbed by access to moisture.
-
2.
Since sodium enhances wetting of cathode carbon, it also allows penetration of electrolyte into zones where secondary reactions occur.13,14
-
3.
The cathode block has direct contact with Al and can then form aluminum carbide at the interface. Al4C3 is electrically conducting, but being a solid material it is dependent on solid-state diffusion and therefore is produced at a low rate. However, the carbide layer can detach from its substrate as the carbonaceous structure changes through intercalation of sodium. Then, aluminum carbide flakes become a particulate impurity that can be entrapped in either the electrolyte or the metal.

Consequences of sodium codeposition in contact with carbonaceous cathode lining; (a) NaOH being formed by reaction with moisture in the air after penetrating the sidewall; (b) condensed Na in spent pot liner rubble after being exposed to moisture.
Also note that, when a zone in the cathode is subject to a very high current density through turbulence, improperly set anodes, cracks in cathode blocks, etc., rather than producing an aluminum alloy, pure sodium gas becomes a localized cathode product. It then combusts as a bright, yellow flame, as illustrated in Fig. 7.

Sodium flare related to a burned-off anode.
Sodium is not often thought of as an impurity in smelters. Those in pot line operations may desire to see higher levels of sodium in the metal produced. High levels of Na are considered to be an indicator of high current efficiency15 generated by the following mechanism.
Cool and stable cells that remain on the lean side of the alumina concentration curve are more likely to have thin layers of cryolite, Na3AlF6, solidify on portions of the surface of the metal pad. Positively charged sodium ions are strongly attracted to the cathode (metal pad), creating a polarization condition, like a boundary layer, at the interface. This polarization drives localized bath chemistry towards the composition of cryolite with a liquidus temperature that is well above that of the rest of the melt. With thin zones of solids buoyed on the surface of the metal, sodium ions are still strongly attracted to the cathode, but the available surface area for Na metal to dissolve in the electrolyte is reduced, effectively concentrating Na in the metal.
Calcium
Calcium in CPC can account for more than 30% of total Ca inputs to the electrolyte. High-pressure water lances are used to cut GPC out of coking shafts or towers prior to sizing and calcination. The hardness of the water leaves a residue of Ca. With the product being referred to as sponge coke, one can easily imagine how the pore structure and surface area of GPC retain calcium impurities as cutting water evaporates.
A fraction of crude oil sources have high total acid number (TAN) values. These contribute substantially to the Ca content of coke. This has been particularly true of some coke sources in China. Poor operation of de-salter units at petrochemical refineries also contribute to high Ca levels in CPC. However, these two conditions are not the norm for the industry (Fig. 8).

Ca in green petroleum coke fractions over and under 1 mm in size.
A modest amount of calcium may also enter via housekeeping activities in a smelter. Pieces of broken concrete, mortar, bricks, refractories, and concrete dust swept from floors all carry small amounts of Ca (as well as Si) into ACM passing through the bath processing plant. In some dry climates where limestone-based sand is commonly found, small amounts of Ca can also enter with dust that settles from the air.
As with sodium, the major input of calcium to the process is with SGA.7 This seldom presents a problem unless the average CaO/Na2O ratio of all alumina used over the course of 6 months or more exceeds 12%. Fortunately, most alumina sources do not exceed this value (Fig. 9).

CaO/Na2O ratios from various alumina refineries.
An average of 4.5–6.5 wt.% CaF2 is commonly found in electrolyte. It is beneficial through enabling lowering of the operating temperature, with the side benefit of reducing the fraction of hard crust,16 while not introducing significant contamination of the metal. However, its concentration is limited because of the impact it has on other physicochemical properties that can harm performance. This includes increasing the specific gravity of the electrolyte, bringing it closer to that of liquid aluminum. A reasonable separation of liquid bath (approx. 2.10 g/cm3) from liquid metal (approx. 2.30 g/cm3)17 is needed to avoid negative consequences such as back-reaction.
The primary sink for Ca entering the process is CaF2 leaving with excess bath generation. This loss rate is directly related to the CaO/Na2O ratio and is most often driven by the Na2O content of SGA.
If the average CaF2 content falls below target, it is easy to add fluorspar so long as impurity inputs such as Si and P are managed. However, if the CaF2 content should rise above 6.5%, difficult and expensive remedies may need to be applied. By closely following CaF2 concentration trends, it may be possible to acquire shipments of alumina that are low in CaO content. Other options include making additions either of Na2CO3 plus AlF3 or of synthetic cryolite to produce pure bath, diluting the CaF2 concentration. Each approach is expensive, especially if excess bath is already being produced.
As the CaF2 content of the electrolyte increases, it will also limit the maximum target for excess AlF3. This is also true for other constituents such as KF, LiF, and MgF2 when they are present in concentrations above de minimis levels. As these impurities increase, electrolyte composition drifts away from what may be considered as “normal” chemistry, such as 5% CaF2 and 11% excess AlF3. An old rule of thumb from the days of Reynolds Metals for complex electrolyte compositions is that, if excess AlF3 plus CaF2, MgF2, and LiF sum to greater than 16%, the process may be entering a zone of concern for stability and sludge deposits. All constituents of the electrolyte must be considered together, not independently.
While a few ppm of Ca are usually found in the metal of reduction cells, excursions of high Ca in metal products have most commonly been traced to cast house operations and alloy materials.
Calcium does not intercalate into cathodes as aggressively as sodium. However, calcium fluoride in bath will carry Ca contamination into newly produced anodes, where it contributes to increased CO2 reactivity and higher amounts of anode dusting in reduction cells.18
Potassium and Radioactivity
For all practical purposes, potassium (K) enters entirely with alumina. The K2O content in most SGA is low, between 4 ppm and 20 ppm. However, some sources in China have been reported to have much higher levels, in the range of 250–625 ppm of K2O equivalent.19
Potassium does offer some significant potential benefits. It lowers the liquidus temperature of the electrolyte and increases the alumina solubility limit. But potassium also causes “large directional swelling of crystallites and grains in baked carbon and graphite artifacts, sometimes resulting in total disintegration” of cathode materials.10 Like sodium, it intercalates into the microstructure of cathode blocks, causing damage when concentrations of KF exceed 5 mol.%. Since potassium can take such a costly and significant toll on cell life, it is generally avoided by most smelters.
As with other impurities, potassium can be found in higher concentration of some bauxite deposits. Metson et al.19 showed concentrations of up to 520 ppm K in alumina from Henan Province of China, resulting in concentrations of up to 4.7% KF in electrolyte. Codeposited impurity levels of K in the bauxite sources of this alumina render the resulting electrolyte unsuitable for commonly used cathode materials. They also render excess bath from these smelters unsuitable for use at other locations.
Another aspect of potassium is that K-40 is a naturally occurring radioactive isotope. This makes bath slightly more radioactive than the background radiation levels of the Earth’s crust.
Other trace elements such as strontium, uranium (typically < 40 ppm), and thorium (typically < 200 ppm) also form fluorides that accumulate in bath. These trace elements are codeposited with bauxite in various parts of the world. However, Th-234 is most likely a natural alpha decay product of U-238.
While radioactivity is often an unsettling topic, the purpose here is not to alarm the reader. Being around granite counters or bunches of bananas exposes a person to more radiation. However, be advised that shipments of excess bath that enter or leave a smelter, or that pass an international border, have been known to set off low-level radiation detection equipment.
As a final note on radioactive impurities, strontium carbonate (SrCO3) has been used as an additive for making accurate determinations of the mass of electrolyte in pots,20 another example of a useful benefit of an impurity.
Lithium
For a few decades, a number of smelters modified their electrolyte chemistry by making regular additions of lithium carbonate (Li2CO3) to attain 2% to 3% LiF content. It was primarily used at smelters with older technology that found lithium carbonate to be an enabler to a more competitive position. Lithium fluoride significantly lowers the electrical resistivity of electrolyte, and it also lowers the liquidus temperature.21 This combination enabled opening the anode–cathode distance and operating at lower temperatures, with reduced metal solubility and higher current efficiencies.
An undesirable side-effect arises from codeposition of Li forming an alloy with Al. Levels of 2–3% LiF place Li concentrations in metal at approximately 20 ppm, well above the acceptable specification limits for many products. Lithium also forms a solid solution with Al, and during solidification it forms small crystals of LiAl that can generate defects in thinly rolled sheets. These effects created the need to use TAC or charcoal decanting to reduce Li in metal products.22
Modern cell technologies have design elements and operational practices that have surpassed performance levels achieved with “lithium chemistry.” This, plus large price increases for Li2CO3, brought its use as an electrolyte additive to an end.
However, concentrations of 40–50 ppm Li are found in some SGA. Li2O is often found in bauxite that is mined around the eastern rim of the Mediterranean Sea, Saudi Arabia, and in Minas Gerais, Brazil. Such alumina sources modestly enhance the conductivity and the liquidus temperature of electrolyte while avoiding the high cost of Li2CO3 additions. Locations that use it may also need to treat some of their metal production to reduce its lithium content prior to casting.
Certain bauxite deposits in China, such as in Henan Province, have produced alumina feedstock to certain smelters, with 400 ppm to 800 ppm of Li causing unusually high LiF content in the electrolyte.19 As with the quote from Aesop, “It is possible to have too much of a good thing.” Concentrations of LiF in the electrolyte in the range of 4% to 9% are essentially in uncharted territory, inviting operational problems. While sales of this lithium-rich bath to other smelters may be tempting, one only need look at the high levels of other contaminants such as potassium, magnesium, or beryllium that may also be found in this material to see that it also is generally unsuitable for use at other smelters.
As noted previously, if the LiF content of the electrolyte is significant, more than 0.2%, you may need to revisit your excess AlF3 target and other parameters of pot control strategy. The best way to estimate the LiF content of the electrolyte is often to closely follow the Li content of pot metal samples.
Magnesium
Magnesium inputs accumulate in the electrolyte as MgF2. However, with the exception of some sources of alumina in parts of Russia and China, magnesium typically is not found at levels of concern in any of the raw materials for the Hall–Héroult process. At some smelters in China, levels of 1–2% MgF2 in electrolyte have been reported.19
Occasionally, magnesium fluoride has been used in combination with CaF2.17,23 MgF2 is more effective at lowering the liquidus temperature of the electrolyte than is CaF2. A few smelters have also added MgO prior to starting new pots in attempts to slow sodium intercalation. The desired effect has not been demonstrated, and this is not a recommended practice, especially for smelters that are not equipped or staffed to deal with complex electrolyte chemistry.
The most recent form of Mg input to the electrolyte has been associated with efforts towards in-house recycling of dross. Not all dross contains Mg, but dross from 5xxx and 6xxx alloys does. There may be 0.5–5% Mg in such alloys, and the impurities tend to concentrate in the dross. If this dross is returned to the smelter for recovery of its Al and Al2O3 content, it can also have a lasting effect on the electrolyte.
An advantage of MgF2 is that it is more easily managed than other fluorides such as CaF2. The partition coefficient for Mg has been estimated at 164:1 (concentration in electrolyte versus concentration in metal), allowing its concentration in the electrolyte to be dissipated relatively quickly by metal production23 when compared with impurities such as Li (310:1), Be (440:1), and Ca (approximately 6800:1). This means that the concentration of MgF2 in the electrolyte will decrease more rapidly over time should a lower concentration target in the electrolyte become desired. It also means that some Mg will be present in all metal produced, but at concentrations that typically are not problematic.
The negative impacts of MgF2 include higher specific gravity of the electrolyte and lower alumina solubility. Changes in the MgF2 content of the electrolyte should also prompt operations to revisit and perhaps change its excess fluoride targets.
Beryllium
Traces of beryllium and lithium are often co-located in bauxite deposits, although this is not universal. There are bauxite deposits in and around the Caribbean and the northeast coast of Australia that have low to moderate concentrations of beryllium and insignificant levels of lithium. Moderate to high levels of beryllium are respectively found in the bauxite deposits of Saudi Arabia, the Balkan Peninsula, Jamaica, and regions of China.
Beryllium is a worker health concern. The World Health Organization has recognized the toxicity of beryllium metal and beryllium compounds. Exposure can lead to allergic reactions that may then lead to chronic beryllium disease, a respiratory disorder. Accordingly, many countries and provinces have adopted strict health guidelines and/or regulations on worker exposure to beryllium.
Fortunately, the form(s) of Be in bath appear to be at least partially soluble in water.8 This may help to explain why the rate of worker health-related incidents in the primary aluminum industry are low.
Beryllium enters the Hall–Héroult process via two pathways: alumina sources from the regions noted above, and excess bath purchased from other smelters that use these alumina sources.8 It tends to concentrate in the electrolyte at approximately 60 times its concentration in SGA.
The most practical approach to reduce the concentration of BeF2 and Na2BeF4 in bath is to use SGA that is very low in beryllium content over long periods. Figure 10 illustrates how Be accumulation and dilution periods compare. Both take multiple years to arrive at a steady-state condition, with dilution taking more time.

Model of accumulation and dilution rates for Be in bath (reprinted from8).
Beryllium has some positive impacts on metal casting. Even a fraction of a ppm of Be in aluminum metal aids casting of 5xxx series, 6xxx series, and foundry alloys by reducing oxidation, enhancing metal yield, and yielding more consistent metallurgical properties. Ultimately, there are limits that must be considered. Many metal products have strict maximum specifications of 1.0 ppm Be. This can be unattainable at smelters with more than 440 ppm of beryllium in bath.
Phosphorus
Phosphorus has been included since the partitioning of P favors accumulation in the electrolyte versus metal.19,24 Although phosphorus does not have a tendency to convert to fluoride, dissolved Na metal could potentially reduce P2O5 to the trivalent state as shown by the following reaction:
When phosphorus enters electrolyte (under electrolysis), a means for cycling between valence states is created that leads to the loss in current efficiency commonly ascribed to it.25,26
It is generally considered that the partition coefficient between electrolyte and metal is approximately 10:119,27 and that 100 ppm of P in electrolyte (approx. 10 ppm in Al) is roughly equivalent to a loss of 1.0% current efficiency.1,28 Accordingly, primary aluminum producers are focused on this impact. Repetitive reduction and oxidation cycles of phosphorus compounds, as in Eq. (7), drive the loss in efficiency.1 This is commonly referred to as a redox reaction.
Phosphorus in various forms enter the Hall–Héroult process with a variety of materials, including SGA, CPC, and AlF3. Ferro-phosphorus additions to the cast iron used to seal anode and cathode assemblies is another source. Phosphorus in various forms may also enter with material streams from cast houses, including bone ash and materials that use phosphate binders.
Once phosphorus enters a pre-bake smelter with modern dry scrubbing systems, it recirculates multiple times, compounding losses in current efficiency. Most oxide and fluoride species of phosphorus have low melting and boiling points when compared with cell temperatures. Thus, various forms of phosphorus in the electrolyte including P2O3 are quite volatile, immediately converting to ultrafine particulate after exiting the bath. These particles are then carried by the exhaust gas stream and are caught on the fabric media filter bags in the dry scrubbers only to be returned to the reduction cells with fluorinated alumina. Its concentration in fluorinated alumina has been measured at one-half to a full order of magnitude higher than what is typical of the SGA.26 Other forms of phosphorus on carbon dust settle on the pot crust only to be recycled with the ACM.
Purging phosphorus from a smelter can be difficult and generally takes time. Phosphorus exits with the metal, through carbon dust skimming (that is not recycled), and via volatile losses, mostly during pot tending activities such as anode changing and metal or bath tapping.
In side-worked and Søderberg cells, including those with point feeders, there is less recirculation of phosphorus. Fume collection rates are generally low, there is no ACM to recirculate, and crust breaking activities are frequent enough that phosphorus (oxide) escapes quickly with other fugitive emissions. It is also removed via skimming out carbon dust accumulations. Less phosphorus is retained in the metal on these types of cells24 (Fig. 11).

Range of phosphorus in metal from various smelters.
The best way to manage phosphorus contamination is to closely control raw materials and material recirculation circuits to minimize total input rates. This can be very challenging. Regular and accurate determinations of P in raw materials and materials that arrive to the bath treatment plant will add some cost and will require proper analytical capability and equipment. Those that follow these inputs will observe that most phosphorus enters with SGA and CPC. Aluminum fluoride and ferrophosphorus additions to cast iron for anode sealing should also be on the list of monitored inputs. Measurements of P flowing into ACM from crucible cleaning, dross recycling, and TAC stations may also be important.
Phosphorus input rates to smelters are often ignored, being in the background of impurity concerns. This changes whenever a large amount of phosphorus enters a smelter over a short period of time. The evidence is that phosphorus excursions in smelters have been related to a variety of one-off events caused by materials.
One such event was traced to CPC impacted by the use of phosphate corrosion inhibitor in the pipes of a petrochemical refinery. The coke producer did not know about the contamination since the phosphorus content of residual tar is not typically reported. There have been substitutions of fluorspar sources at AlF3 producers that have been high in phosphorus. There have also been cases of carryover of phosphorus from bone ash from cast houses that then contaminated ACM. Such issues claim attention by causing acute losses in current efficiency and production. Nonetheless, every kilogram of P that enters the Hall–Héroult process takes some toll, no matter if the entry pathway is insidious or acute.
Conclusions
What has been presented here is only information until it is used. So, what may be done about the various impurities and conditions that have been reviewed?
Note that excessive levels of contaminants, including Ca, K, Li, and P, are beyond simple tips to resolve dire situations. Pre-planning, monitoring, and diligence are the most useful and sometimes the least utilized tools for prevention. Responding to problems implies that actions are being taken later than they should have.
Identification of the impurities and processes that must be kept under control comes first.
-
Some items identified during planning are conditional, e.g., “If we purchase alumina or bath originating from source X, we shall take certain, pre-specified steps.”
-
Others will be ongoing, such as monitoring the CaO/Na2O ratio of alumina receipts over time to track against %CaF2 in electrolyte.
-
A few fall under the category of diligence: confirmation of P2O5 content in alumina and AlF3, or phosphorus content of CPC, with any new supplier, monitored over a period of time followed by spot checks of incoming materials.
-
Some will require looking over the fence, such as monitoring of materials passing from the cast house into ACM.
-
Many will require “looking in the mirror, rather than outside the window” to regularly question: if you have set your strategy correctly, if all who should receive communications about various impurities are truly being kept in the loop, and if additional resources, education, or outside help is needed.
These are all valid starting points to begin planning of corrective actions or to face the challenges that may come. Tying it all together with clear organization and regular communications is what may be needed most.
Need for outside assistance may include analytical procedures. Not all impurities are easy to measure with accuracy. For example, when using XRF analysis for phosphorus determination, it is important to be aware of the dominance of the spectral peak of Al in relation to the weaker spectral peak of P. It is always a good practice to meet with the laboratory analyst to discuss the detection limits of the procedures and instrumentation and anticipated concentration ranges to ensure that proper standards are available and that actual results will be within the gauge capability of the analytical instruments. Beryllium analysis requires a specialized procedure of digestion for samples of alumina, bath, or AlF3.
Some items are easier. It may be difficult to measure the Li content of Al2O3 or bath at low concentrations. Using published literature and the partition coefficient estimates presented above can make it possible to accurately estimate these by following Li in pot metal samples.
The following is a point-by-point review of what has been discussed in this article:
-
(1)
Impurities are needed to maintain the electrolyte in its optimum condition.
-
a.
Are electrolyte chemistry targets set using rigorous analytical processes, or are some, such as %CaF2, %KF, and %LiF, determined by whichever raw material has the best price?
-
b.
CaF2 content and the content of other fluorides may vary over time. Is the excess AlF3 concentration adjusted to compensate? If the CaF2 content begins to drift, are tools that “look ahead” and pre-established maximum limits used to change raw material sourcing?
-
c.
Have maximum limits been established for K2O or Be content that cannot be purchased without a full review by management and technical resources?
-
d.
If fluorspar is being used to increase the CaF2 content, is it added slowly to minimize the impact of Si and P contamination?
-
a.
-
(2)
As an impurity level climbs, are other actions taken to compensate?
-
a.
The most common examples of this are related to impurities such as Fe or Si that contaminate the metal. However, there are choices that can be made with impurities that affect bath chemistry. For example, the excess fluoride content target may be reduced as the concentrations of other fluorides increase.
-
b.
Calcium enters through multiple pathways. The CaO/Na2O ratio monitors the largest input, alumina. Changing CPC sourcing may be an option for reducing Ca input. Establishing work practices to keep chunks of concrete and concrete dust out of ACM makes sense. Even the choice of a different alumina source with higher Na2O content may be less expensive than straightforward dilution options for high CaF2 content.
-
c.
Beryllium enters with alumina or with bath purchased from other smelters. Are the more and less desirable sources known in advance? If beryllium content may increase, are industrial hygiene professionals among the first to be informed?
-
d.
If K or Li are of concern, have processes been put into place to check all likely sources of alumina or bath before the time of need?
-
e.
If dross from 5xxx and/or 6xxx series is on site and any streams of dross are being added to ACM, measures as simple as training and auditing can help to prevent significant contamination of the electrolyte.
-
a.
-
(3)
If your situation is one of complex electrolyte chemistry, with significant amounts of LiF or MgF2, or even constantly changing concentrations of CaF2, are resources available to keep electrolysis on the proper course?
-
a.
Should the concentrations of LiF, MgF2, KF, or CaF2 change, the liquidus temperature of the electrolyte will also change. Will operational expectations for average bath temperature change too, or is superheat and side ledge protection put at risk? Will the definitions for hot and cold pots be adjusted?
-
b.
If superheat probes are used, will its reference curves be recalibrated to adapt to changes in electrolyte constituents that it cannot measure?
-
a.
-
(4)
Is it truly known what arrives at the bath treatment plant from all sources?
-
a.
An example of bone ash in cast houses was given in the text. Does pre-planning include a walk-through of the cast house with knowledgeable persons? Are phosphate-bonded filter boxes added to dross? Has additional salt been added for fluxing? Will the metal product mix change, generating more dross containing certain impurities?
-
b.
If the product mix is changing, will there be more, or less, need to reduce levels of sodium in metal by using metal treatment processes?
-
c.
Are processes and materials audited to ensure that pot basement clean-out has methods, expectations, and tools for removing chunks of concrete?
-
d.
If floor sweepings are disposed of, are sweeping operations ever audited to ensure that the sweeper catch is not returning to the bath treatment plant and the ACM?
-
a.
-
(5)
Are input rates of phosphorus in raw materials, anode castings, and use of phosphate-bonded refractories tightly controlled and monitored?
-
a.
Phosphorus enters with many materials. Are adequate safeguards in place on each source to prevent acute or chronic problems? Confirmation testing, regular monitoring of raw materials, and new supplier qualifications all fall in the area of prevention. Hazard familiarization, training, and communication with procurement do as well.
-
b.
Trend analysis on average concentrations of P in pot metal samples can help to guide additional actions. Note that phosphorus is volatile enough that using anything but metal samples from pots may lead one astray.
-
a.
We hope that this document serves as an enduring reference on the nature of impurities in Hall–Héroult baths and that this summary serves to guide your path forward.
References
E.W. Thisted, Electrochemical Properties of Phosphorus Compounds in Fluoride Melts, PhD thesis, (Norwegian University of Science and Technology, 2003)
G.T. Holmes, Light Metals 1995, ed. J.W Evans, (Minerals, Metals & Materials Society, 1995), pp. 371–373
W.H. Tingle, J. Petit and W.B. Frank, Proceedings of the Symposium on Molten salts, Electrochemical Society Meeting, Washington, D.C., May 2–7, 1976
K. Grjotheim, Contribution to the Theory of the Aluminum Electrolysis, PhD thesis, (Norwegian University of Science and Technology, 1956)
R.J. Snow, and B.J. Welch, R.J. Snow, and B.J. Welch, J. Electrochem. Soc. 115, 1170. (1968).
A.S. Dworkin, H.R. Bronstein, and M.A. Bredig, A.S. Dworkin, H.R. Bronstein, and M.A. Bredig, Phys. Chem. 66, 572. (1962).
S. J. Lindsay, Proceedings of the 9th Australasian Aluminium Smelting Technology Conference, 2007, pp. 5-22
S. Lindsay and C. Dobbs, Light Metals 2006, ed. T.J. Galloway (Minerals, Metals & Materials Society, 2006), pp. 95–100
K. Grjotheim, and B. Welch, Aluminium Smelter Technology, 2nd edn. (Aluminum Verlag, Düsseldorf, 1988), pp 57–63
M. Sørlie, H. Øye, Cathodes in Aluminum Electrolysis, 3rd ed. (Aluminum Verlag, 2010), pp. 232–234, pp. 520–521
W.K. Fischer and R.C. Perruchoud, Light Metals 1991, ed. E. Rooy (Minerals, Metals & Materials Society, 1991), pp. 721–724
J.F. Grandfield, Fundamentals of Aluminum Metallurgy—Production, Processing and Applications, ed. Roger Lumley (Cambridge: Woodhead Publishing Ltd., 2011), section 5.10.4
M.B. Dell, Light Metals 1985, ed. H.A. Bohner (Warrendale, Pa.: Metallurgical Society of AIME, 1985), pp. 957–965
P. Rafiei, F. Hiltmann, M. Hyland, B. James and B. Welch, Light Metals 2001, ed. J.L. Anjier (Minerals, Metals & Materials Society, 2001), pp. 747–752
L. Dion, L. Kiss, P. Chartrand, G. Dufour and F. Laflamme, Light Metals 2013, ed. B.A. Sadler (Wiley, 2013), pp. 741–746
M. Graham and B. Pruitt, R&D for Industry: A Century of Technical Innovation at Alcoa, (New York: Cambridge University Press, 1990, p. 47
N. E. Richards, Alumina in Smelting, (11th International Course on Process Metallurgy of Aluminium, 1992), Chapter 10, p.3
K. Khaji and M. Al-Qassemi, Light Metals 2015, ed. M. Hyland (Springer, Cham, 2015), pp. 1135–1140
J.B. Metson, D.S. Wong, J.H. Hung and M.P. Taylor, Light Metals 2013, ed. B.A. Sadler, (The Minerals, Metals & Materials Series. Springer, Cham, 2013), pp. 9–13
M. Iffert, Aluminum Smelting Cell Control and Optimisation, PhD thesis (University of New South Wales, 2008), pp. 124–133
A.T. Tabereaux, T.R. Alcorn and L. Trembley, Light Metals 1993, ed. S.K. Das (Minerals, Metals & Materials Society, 1993), pp. 221–226
A. Abbe, Light Metals 2009, ed. G. Bearne (The Minerals, Metals & Materials Society, 2009), pp. 529-532
W. Haupin, Properties of Bath, (11th International Course on Process Metallurgy of Aluminium, 1992), Chapter 5, p.15
E. Haugland, G.M. Haarberg, E. Thisted and J. Thonstad, Light Metals 2001, ed. J.L. Anjier (Minerals, Metals & Materials Society, 2001), pp. 549–553
P.A. Solli, T. Haarberg, T. Eggen, E. Skybakmoen and Å. Sterten, Light Metals 1994, ed. U. Mannweiler (The Minerals, Metals & Materials Society, 1994), pp. 195–203
J.A. Al-Mejali, G.M. Haarberg, N. Bensalah, B.A. Benkahla and H.P. Lange, Light Metals 2016, ed. E. Williams (Springer, Cham, 2016), pp. 389–394
A. Tabereaux, Phosphorus Impurity in Aluminum Electrolysis Cells, (TMS Course - Smelter Grade Alumina from the Smelting Perspective, 2004), Charlotte, N.C., Chapter 2, pp. 1–13
E. Sturm and G. Wedde, Light Metals 1998, ed. B. Welch, (Minerals, Metals & Materials Society, 1998), pp. 235–240
Acknowledgements
We thank Nik Winjum and Les Edwards for their support in the preparation of this article. We are also grateful to Merino, Margarita R. (Ph.D. – Florida State University) for her encouragement, dedication, and support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lindsay, S.J., Welch, B.J. A Review: Understanding the Science and the Impacts of Impurities upon the Electrolytic Bath of Hall–Héroult Reduction Cells. JOM 73, 1196–1209 (2021). https://doi.org/10.1007/s11837-021-04572-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11837-021-04572-7