
Abstract
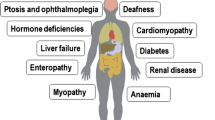
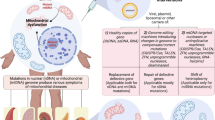
Mitochondrial disorders, namely defects of fatty acid oxidation, defects of pyruvate metabolism and defects of the respiratory chain are heterogenous in clinical picture and in response to therapeutic attempts. Defects of fatty acid metabolism are amenable to therapy by dietary means, carnitine substitution and in some cases with vitamins. Defects in pyruvate metabolism do not respond to therapy except in some special cases. Therapeutic attempts include dietary measures, vitamins as coenzyme precursors. Defects in the respiratory chain appear to respond to treatment only in exceptional cases. Evaluation of treatment effects appears to be singularly difficult. General measures that can be of benefit to different defects are discussed.
Similar content being viewed by others

References
Arts, W. F. M., Scholte, H. R., Bogaard, J. M., Kerrebijn, K. F. and Luyt-Houwen, I. E. M. NADH-CoQ reductase deficient myopathy: successful treatment with riboflavine.Lancet 2 (1983) 581–582
Atkin, B. M., Buist, N. R. M., Utter, M. F., Leiter, A. B. and Banker, B. Q. Pyruvate carboxylase deficiency and lactic acidosis in a retarded child without Leigh's disease.Pediatr. Res. 13 (1979) 109–116
Baal, M. G., Gabreëls, F. J. M., Renier, W. O., Hommes, F. A., Gijsbers, Th. H. J., Lamers, K. J. B. and Kok, J. C. N. A patient with pyruvate carboxylase deficiency in the liver: treatment with aspartic acid and thiamine.Dev. Med. Child Neurol. 23 (1981) 521–530
Bank, W. J., Di Mauro, S., Bonilla, E., Capuzzi, D. M. and Rowland, L. P. A disorder of muscle lipid metabolism and myoglobinuria.N. Engl. J. Med. 292 (1975) 443–449
v. Biervliet, J. P. G. M., Bruinvis, L., v. d. Heiden, C., Ketting, D., Wadman, S. K., Willemse, J. L. and Monnens, L. A. H. Report of a patient with severe, chronic lactic acidaemia and pyruvate carboxylase deficiency.Dev. Med. Child Neurol. 19 (1977) 392–401
Blass, J. P., Kark, R. A. P. and Engel, W. K. Clinical studies of a patient with pyruvate decarboxylase deficiency.Arch. Neurol. 25 (1971) 449–460
Bougnères, P. F., Saudubray, J. M., Marsac, C., Bernard, O., Odièvre, M. and Girard, J. Fasting hypoglycemia resulting from hepatic carnitine palmitoyl transferase deficiency.J. Pediatr. 98 (1981) 742–746
Brunette, M. G., Delvin, E., Hazel, B. and Scriver, C. R. Thiamine responsive lactic acidosis in a patient with deficient lowK m pyruvate carboxylase activity in liver.Pediatrics 50 (1972) 702–711
Cederbaum, S. D., Blass, J. P., Minkoff, N., Brown, W. J., Cotton, M. E. and Harris, S. H. Sensitivity to carbohydrate in a patient with familial intermittent lactic acidosis and PDH deficiency.Pediatr. Res. 10 (1976) 713–720
Crabb, D. W., Yount, E. A. and Harris, R. A. The metabolic effects of dichloroacetate.Metabolism 30 (1981) 1024–1039
Cumming, W. J. K., Hardy, M., Hudgson, P. and Walls, J. Carnitine palmityl transferase deficiency.J. Neurol. Sci. 30 (1976) 247–258
Di Donato, S., Rimoldi, M., Moise, A., Bertagnoglio, B. and Uzil, G. Fatal ataxic encephalopathy and carnitine acetyl transferase deficiency a functional defect of pyruvate oxidation?Neurology 29 (1979) 1578–1583
Eleff, S., Kennaway, N. G., Buist, N. R. M., Darley-Usmar, V. M., Capaldi, R. A., Bank, W. J. and Chance, B.31P NMR study of improvement in oxidative phosphorylation by vitamins K3 and C in a patient with a defect in electron transport at complex III in skeletal muscle.Proc. Natl. Acad. Sci. USA 81 (1984) 3529–3533
French, J. H., Sherard, E. S., Lubell, H., Brotz, M. and Moore, C. L. Trichopoliodystrophy. I. Report of a case and biochemical studies.Arch. Neurol. 26 (1972) 229–244
Gregersen, N., Christensen, M. F., Christensen, E. and Kolvraa, S. Riboflavine responsive multiple acyl-CoA dehydrogenation deficiency.Acta Pediatr. Scand. 75 (1986) 676–681
Harpey, J. P., Charpentier, C. and Coudé, M. Methylene blue for riboflavin-unresponsive glutaric aciduria type II.Lancet 1 (1986) 391
Hayes, D. J., Hilton-Jones, D., Arnold, D. L., Galloway, G., Styles, P., Duncan, J. and Radda, G. K. A mitochondrial encephalomyopathy. A combined31P magnetic resonance and biochemical investigation.J. Neurol. Sci. 71 (1985) 105–118
Hommes, F. A., Polman, H. A. and Reesink, J. D. Leigh's encephalomyelopathy: an inborn error in gluconeogenesis.Arch. Dis. Child. 43 (1968) 423–426
Jusko, W. J. and Levy, G. Absorption, protein binding and elimination of riboflavin. In Rivlin, R. S. (ed.)Riboflavin, Plenum Press, New York, 1975, pp. 99–152
Maesaka, H., Komiya, K., Misugi, K. and Tada, K. Hyperalaninemia, hyperpyruvicemia and lactic acidosis due to pyruvate carboxylase deficiency of the liver; treatment with thiamine and lipoic acid.Eur. J. Pediatr. 122 (1976) 159–168
Matalon, R., Stumpf, D. A., Michals, K., Hart, R. D., Parks, J. K. and Goodman, S. I. Lipoamide dehydrogenase deficiency with primary lactic acidosis: favourable response to treatment with oral lipoic acid.J. Pediatr. 104 (1984) 65–69
McCormick, K., Viscardi, R. M., Robinson, B. and Heininger, J. Partial pyruvate decarboxylase deficiency with profound lactic acidosis and hyperammonemia: response to dichloroacetate and benzoate.Am. J. Med. Genet. 22 (1985) 291–299
McKay, N., Petrova-Benedict, R., Thoene, J., Bergen, B., Wilson, W. and Robinson, B. Lacticacidaemia due to pyruvate dehydrogenase deficiency, with evidence of protein polymorphism in the α-subunit of the enzyme.Eur. J. Pediatr. 144 (1986) 445–450
Mooy, P. D., Przyrembel, H., Giesberts, M. A. H., Scholte, H. R., Blom, W. and van Gelderen, H. H. Glutaric aciduria type II: treatment with riboflavin, carnitine and insulin.Eur. J. Pediatr. 143 (1984) 92–95
Morgan-Hughes, J. A., Hayes, D. J., Clark, J. B. and Cooper, J. M. Mitochondrial myopathies. Results of exploratory therapeutic trials. In Folkers, K. and Yamamura, Y. (eds.),Biochemical and Clinical Aspects of Coenzyme Q. Vol. 4, Elsevier Ssience Publishers, Amsterdam, 1984, pp. 417–424
Munnich, A., Saudubray, J. M., Taylor, J., Charpentier, C., Marsac, J., Rocchiccioli, F., Amedee-Manesme, O., Coudé, F. X., Frézal, J. and Robinson, B. H. Congenital lactic acidosis, α-ketoglutaric aciduria and variant form of maple syrup urine disease due to a single enzyme defect: dihydrolipoyl dehydrogenase deficiency.Acta Paediatr. Scand. 71 (1982) 167–171
Ogasahara, S., Nishikawa, Y., Yorifuji, S., Soga, F., Nakamura, Y., Takahashi, M., Hashimoto, S., Kono, N. and Tarui, S. Treatment of Kearns-Sayre syndrome with coenzyme Q 10.Neurology 36 (1986) 45–53
Patten, B. M., Wood, J. M., Harati, Y., Hefferan, P. and Howell, R. R. Familial recurrent rhabdomyolysis due to carnitine palmityltransferase deficiency.Am. J. Med. 67 (1979) 167–171
Przyrembel, H. Carnitin: Bedeutung für Klinik, Diagnostik, Therapie.Akt. Endokr. Stoffw. 7 (1986) 9–17
Reza, M. J., Kar, N. C., Pearson, C. M. and Kark, R. A. P. Recurrent myoglobinuria due to muscle carnitine palmityltransferase deficiency.Ann. Intern. Med. 88 (1978) 610–615
Robinson, B. H. and Sherwood, W. G. Pyruvate dehydrogenase phosphatase deficiency: a cause of congenital chronic lactic acidosis in infancy.Pediatr. Res. 9 (1975) 935–939
Robinson, B. H., Taylor, J., Kahler, S. G. and Kirkman, H. N. Lactic acidemia, neurologic deterioration and carbohydrate dependance in a girl with dihydrolipoyl dehydrogenase deficiency.Eur. J. Pediatr. 136 (1981) 35–39
Robinson, B. H., Taylor, J., Francois, B., Beaudet, A. L. and Peterson, D. F. Lacticacidosis, neurological deterioration and compromised cellular pyruvate oxidation due to a defect in the reoxidation of cytoplasmically generated NADH.Eur. J. Pediatr. 140 (1983) 98–101
Roe, C. R., Millington, D. S., Maltby, D. A., Bohan, T. P., Kahler, S. G. and Chalmers, R. A. Diagnostic and therapeutic implications of medium-chain acylcarnitines in the medium-chain acyl-CoA dehydrogenase deficiency.Pediatr. Res. 19 (1985) 459–466
Snyder, T. M., Little, B. W., Roman-Campos, G. and McQuillen, J. B. Successful treatment of familial idiopathic lipid storage myopathy withl-carnitine and modified lipid diet.Neurology 32 (1982) 1106–1115
Stacpoole, P. W., Harman, E. M., Curry, S. H., Baumgartner, T. G. and Misbin, R. I. Treatment of lactic acidosis with dichloroacetate.N. Engl. J. Med. 309 (1983) 390–396
de Visser, M., Scholte, H. R., Schutgens, R. B. H., Bolhuis, P. A., Luyt-Houwen, I. E. M., Vaandrager-Verduin, M. H. M., Veder, H. A. and Oey, P. L. Riboflavin-responsive lipid-storage myopathy and glutaric aciduria type II of early adult onset.Neurology 36 (1986) 367–372
de Vivo, D. C., Haymond, M. W., Leckie, M. P., Bussmann, Y. L., McDougal, D. B. and Pagliara, A. S. The clinical and biochemical implications of pyruvate carboxylase deficiency.J. Clin. Endocrinol. Metab. 45 (1977) 1281–1296
Warner, A. and Vaziri, N. D. Treatment of lactic acidosis.South. Med. J. 74 (1981) 841–847
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Przyrembel, H. Therapy of mitochondrial disorders. J Inherit Metab Dis 10 (Suppl 1), 129–146 (1987). https://doi.org/10.1007/BF01812853
Issue Date:
DOI: https://doi.org/10.1007/BF01812853