
Abstract
Key message
Genome-wide association analysis identified 61 SNP markers for canopy wilting, which likely tagged 51 different loci. Based on the allelic effects of the significant SNPs, the slowest and fastest wilting genotypes were identified.
Abstract
Drought stress is a major global constraint for crop production, and slow canopy wilting is a promising trait for improving drought tolerance. The objective of this study was to identify genetic loci associated with canopy wilting and to confirm those loci with previously reported canopy wilting QTLs. A panel of 373 maturity group (MG) IV soybean genotypes was grown in four environments to evaluate canopy wilting. Statistical analysis of phenotype indicated wide variation for the trait, with significant effects of genotype (G), environment (E), and G × E interaction. Over 42,000 SNP markers were obtained from the Illumina Infinium SoySNP50K iSelect SNP Beadchip. After filtration for quality control, 31,260 SNPs with a minor allele frequency (MAF) ≥5% were used for association mapping using the Fixed and random model Circulating Probability Unification (FarmCPU) model. There were 61 environment-specific significant SNP-canopy wilting associations, and 21 SNPs that associated with canopy wilting in more than one environment. There were 34 significant SNPs associated with canopy wilting when averaged across environments. Together, these SNPs tagged 23 putative loci associated with canopy wilting. Six of the putative loci were located within previously reported chromosomal regions that were associated with canopy wilting through bi-parental mapping. Several significant SNPs were located within a gene or very close to genes that had a reported biological connection to transpiration or water transport. Favorable alleles from significant SNPs may be an important resource for pyramiding genes to improve drought tolerance and for identifying parental genotypes for use in breeding programs.
Similar content being viewed by others
Introduction
Soybean [Glycine max (L.) Merr.] is among the most widely grown crops in the world and is valuable because of its high oil and protein concentrations. The United States is the leading soybean-producing country, followed by Brazil, Argentina, and China (USDA-FAS 2016). Over the last 60 years, there has been an increasing trend in soybean yield in the US due to improvement in genetic and cultural practices (Irwin and Good 2015). Fox et al. (2013) reported that soybean breeding produced more than 500 cultivars over the last 60 years in North America that contributed to a 25% increase in yield.
Drought occurrence represents a severe abiotic stress and causes a reduction in soybean productivity in rain-fed areas. Drought adversely affects soybean yield to some degree at most developmental stages, particularly, during reproductive development (Oya et al. 2004). Drought is a major cause in the variation of soybean yield from year to year (Zipper et al. 2016) and is projected to be more intense with global climate change. Genetic improvement of soybean for drought tolerance is a cost-effective approach to stabilize yield for rain-fed areas.
Slow canopy wilting is a promising trait for improving drought tolerance. Screening of exotic germplasm for drought tolerance in North Carolina identified several slow-wilting genotypes, including PI 416937 and PI 471938 (Carter et al. 1999, 2006). Several mechanisms are likely to be responsible for slower canopy wilting. Sinclair et al. (2008) reported that slow-wilting in PI416937 was mainly associated with lower leaf hydraulic conductance for transpiration rate under high vapor pressure deficit (VPD). Devi and Sinclair (2013) reported that slow-wilting in PI 471938 was associated with a lower osmotic potential that helped to maintain a greater leaf turgor pressure. Slow-wilting may also be associated with maintaining greater leaf turgor, transpiration, and CO2 exchange rates during drought conditions (Carter et al. 2006; Fletcher et al. 2007; Sadok and Sinclair 2009; Sloane et al. 1990). King et al. (2009) and Ries et al. (2012) determined that slow-wilting was due to the conservation of soil moisture when soil moisture was plentiful. The conserved soil moisture could then be used when soil moisture in fast-wilting genotypes had been depleted.
Drought tolerance is complex and is controlled by a number of genes throughout the genome, each with minor effects and interacting with the environment (Blum 2011; Tuberosa et al. 2007). Under water-limited environments, crop performance can be improved by selecting and pyramiding favorable alleles associated with drought-tolerant related traits into elite cultivars (Blum, 2005). Various genomic approaches have been used to dissect genetic control of drought stress tolerance (Tuberosa et al. 2007). Quantitative trait loci (QTL) identification using molecular markers is one way to dissect the traits associated with drought tolerance (Dixit et al. 2014). Several different mapping populations have been used for QTL mapping of canopy wilting, which identified several genomic regions associated with canopy wilting variation. Charlson et al. (2009) identified four QTLs for canopy wilting on chromosomes Gm08, Gm13, Gm14, and Gm17 using a mapping population of 93 RILs (KS4895 and Jackson). Du et al. (2009) used a mapping population of 184 RILs from the cross of Kefeng1 and Nannong1138-2 to identify two QTLs for canopy wilting that were present on Gm8 and Gm20. Abdel-Haleem et al. (2012) identified seven QTLs for canopy wilting on Gm02, Gm04, Gm05, Gm12, Gm14, Gm17, and Gm19 using a mapping population of 150 RILs (Benning and PI 416937). Hwang et al. (2015) used the results of QTLs for wilting from five mapping populations to identify clusters of eight QTLs that were present in at least two populations, and a meta-analysis of these eight clusters identified nine meta-QTLs in eight chromosomal regions (Hwang et al. 2016).
Genome-wide association studies (GWAS) have emerged as a powerful tool to map and unravel complex trait variation down to the sequence level and to identify the genes associated with those traits (Nordborg et al. 2002; Zhu et al. 2008). The USDA soybean germplasm collection has been genotyped with the SoySNP50K iSelect Beadchip, which has allowed characterization of soybean genetic diversity, linkage disequilibrium, and the construction of high resolution linkage maps (Song et al. 2013). Recently, several GWAS in soybean identified significant SNP markers associated with seed protein and oil concentrations (Hwang et al. 2014), carotenoids (Dhanapal et al. 2015a), δ13C ratio (Dhanapal et al. 2015b), agronomic traits (Wen et al. 2014), and ureide concentrations (Ray et al. 2015).
In the present research, 42,509 SNP markers (www.soybase.org) were utilized for GWAS of canopy wilting on a panel of 373 diverse MG IV accessions. The objectives of this study were to explore the genetic variation of canopy wilting present within this panel, to identify significant SNPs associated with canopy wilting, and to confirm those SNPs with previously reported chromosomal regions associated with canopy wilting variations.
Materials and methods
Field experiments
Field experiments were conducted in four environments including the Pine Tree Research Station, AR (35°7′N, 90°55′W) in 2016 (PT-16) on a Calloway silt loam, Rohwer Research Station, AR (33°48′N, 91°17′W) in 2016 (RO-16) on a Sharkey silty clay, Salina, KS (38°480′N, 97°36′W) in 2015 (SA-15) and 2016 (SA-16) on a Hord silt loam. The experiments were sown on 2 June 2016 (PT-16), 23 May 2016 (RO-16), 9 June 2015 (SA-15), and 15 June 2016 (SA-16). The seeding density for all experiments was 32 m−2. For PT-16 and RO-16, plots were sown with a drill (19-cm row spacing) and were 1.52-m wide and 4.57-m long. At Salina for both years, plots consisted of two rows, 0.76 m apart that were 3.65 m in length.
The experimental design at each environment was a randomized complete block with two replications. Along with 373 maturity group (MG) 4 accessions, two check genotypes, slow-wilting (PI 416937) and fast-wilting (A5959), were evaluated in each environment. PI 416937 and A5959 were confirmed as slow and fast-wilting in the previous research (Hwang et al. 2015, 2016; King et al. 2009). These 373 accessions were obtained from the Soybean Germplasm Collection, USDA-ARS based on GRIN (Germplasm Resources Information Network, www.ars-grin.gov) data as reported by Dhanapal et al. (2015b). These accessions originated from 11 different nations, which increased the genetic diversity.
Phenotypic evaluations and descriptive statistics
Phenotypic evaluation of canopy wilting was scored using a visual rating based on a scale from 0 (no wilting) to 100 (plant death) (King et al. 2009). Canopy wilting was rated two times at PT-16, and three times each at RO-16, SA-15, and SA-16 environments within 2 h of solar noon under a clear sky. For any given rating date, all MG 4 accessions were at approximately the same growth stage, while the two slow-wilting check entries were at a slightly earlier stage. The previous research found that genotypic ranking for wilting was similar between years over a wide range of growth stages and over stress conditions that were mild to severe (King et al. 2009) and that there was little or no discernible differences in wilting due to growth stages prior to R6. Because of a dynamic plant/environment interaction, canopy wilting scores must be taken when conditions are favorable to differentiate genotypes for wilting. For the four environments in this study, conditions favorable for canopy wilting occurred between late vegetative development to R4 across all rating dates. For each environment, the average of all the canopy wilting scores per plot was determined for further analysis. Genotype was treated as a fixed effect and replication within the environment was considered as a random effect. Descriptive statistics and Pearson correlation analysis, for the average canopy wilting scores for each environment were computed using the PROC UNIVARIATE and PROC CORR procedures (α = 0.05) of SAS version 9.4 (SAS, Institute 2013), respectively. For analysis of variance (ANOVA), the PROC MIXED procedure (α = 0.05) of SAS 9.4 was used with a model as suggested by Bondari (2003). The model for the phenotypic trait was \( y_{ijk} = \mu + G_{i} + E_{j} + \left( {\text{GE}} \right)_{ij} + B_{{k\left( {ij} \right)}} + \varepsilon_{ijk} , \) where \( \mu \) is the total mean, \( G_{i} \) is the genotypic effect of the ith genotype, \( E_{j} \) is the effect of the jth environment, \( \left( {\text{GE}} \right)_{ij} \) is the interaction effect between the ith genotype and the jth environment, \( B_{{k\left( {ij} \right)}} \) is the effect of replication within the jth environment, and \( \varepsilon_{ijk} \) is a random error following \( N\left( {0, \sigma_{e}^{2} } \right) \).
Broad sense heritability on an entry-mean basis was calculated as \( H^{2} = \sigma_{G}^{2} / \left( {\sigma_{G}^{2} + \left( {\frac{{\sigma_{\text{GE}}^{2} }}{k}} \right) + \left( {\frac{{\sigma_{\varepsilon }^{2} }}{rk}} \right)} \right), \) where \( \sigma_{G}^{2} \) is the genotypic variance, \( \sigma_{\text{GE}}^{2} \) is the genotype by environment variance, \( \sigma_{\varepsilon }^{2} \) is the residual variance, k is the number of environments, and r is the number of replications. The PROC VARCOMP of SAS 9.4 with the REML method (restricted maximum likelihood estimation) was used to estimate the above variance components. To reduce the environmental variation, the Best Linear Unbiased Prediction (BLUP) values for each independent environment and across all environments were estimated using the PROC MIXED procedure, and these values were then used in GWAS analysis.
Genotyping and quality control
Marker data available from Soybase (www.soybase.org) provided data on 42,509 SNP markers for all 373 genotypes. There were 31,260 polymorphic markers after performing quality control checks by eliminating monomorphic markers, markers with minor allele frequency (MAF) <5%, and markers with a missing rate higher than 10%. Imputation of remaining marker data was applied using an LD-kNNi method, which is based on a k-nearest-neighbor genotype (Money et al. 2015). After filtration and imputation, the remaining 31,260 SNPs were used for association testing to identify significant SNPs.
Linkage disequilibrium estimation
Pairwise linkage disequilibrium (LD) between markers was measured by squared correlation coefficients (r 2) of alleles in the TASSEL 5.0 software (Bradbury et al. 2007). To understand the difference in recombination rate, LD was calculated separately for euchromatic and heterochromatic regions. Information of physical distance of euchromatic and heterochromatic regions for each chromosome was obtained from Soybase (www.soybase.org). Nonlinear regression curves, as described by Hill and Weir (1988), were used to estimate the LD decay with distance. The decay rate of LD was determined as the physical distance between markers where the average r 2 dropped to half its maximum value.
Genome-wide association analysis
Population stratification can induce false-positives in GWAS. There are a number of statistical models that effectively control these false-positives by incorporating population structure and kinship among genotypes. The most commonly used model for association analysis is the mixed linear model (MLM) that accounts for the family relatedness and population structure (Yu et al. 2006; Zhang et al. 2010). Incorporation of population structure and family relatedness in the MLM models adjusts association tests to control false-positives; however, these adjustments also compromise true positives (Liu et al. 2016). Hence, these models can induce false-negatives due to over-fitting of the model to a degree where potentially important associations can be missed. Fixed and random model Circulating Probability Unification (FarmCPU) effectively corrects false-positives without compromising true positives (Liu et al. 2016). Both MLM and FarmCPU models were compared using average canopy wilting across all environments and results were evaluated based on quantile–quantile (Q–Q) plots. Based on results as described subsequently, the FarmCPU model was chosen for the association analysis reported herein.
In FarmCPU, Multiple Loci Linear Mixed Model (MLMM) is divided into two parts: a Fixed Effect Model (FEM) and a Random Effect Model (REM) which are used iteratively (Liu et al. 2016). To avoid model over-fitting, REM estimates the multiple associated markers that are used to obtain kinship. The FEM tests markers, one at a time, and kinship from REM as covariates to control false-positives and false-negatives. At each iteration, P values of testing markers and multiple associated markers are unified.
A threshold value (−Log10 P ≥ 3.5), which is equivalent to a P value ≤ 0.0003, was used to declare a significant association of SNPs with canopy wilting. This threshold value is more stringent than that reported in other soybean GWAS studies (Dhanapal et al. 2015a, b; Hao et al. 2012; Hwang et al. 2014; Zhang et al. 2015). To identify the common significant SNPs present in more than one environment, a threshold value of P ≤ 0.05 was used, but only if the representative SNP had an association of P ≤ 0.0003 in a second environment.
Genetic merit for each accession was determined using genomic best linear unbiased prediction (gBLUP), which utilizes genomic-relationship matrix and phenotype data (Clark and van der Werf 2013; Zhang et al. 2007) and breeding values for genotypes. The gBLUP values were calculated in a GAPIT program (Lipka et al. 2012). The breeding value of each accession was calculated from the allelic effects of all significant SNPs. The allelic effect was calculated by taking a difference in mean canopy wilting between genotypes with the major allele and those with the minor allele. Alleles from either the major class or minor class were considered as favorable if they were associated with a reduction in the canopy wilting. To estimate the breeding value for each accession, the absolute value of the allelic effect of each significant SNP was considered as a negative value if an accession had a favorable allele of a significant SNP at that location, (i.e., if the allelic effect decreased canopy wilting). Otherwise, if the allelic effect was unfavorable (i.e., increased canopy wilting), the allelic value for an SNP was considered as a positive value. All positive and negative allelic values were summed to estimate the breeding value of each accession.
Candidate gene identification
Significant SNPs at level of −Log10 P ≥ 3.5 were used to identify the candidate genes in each environment and across all environments. Candidate genes, their associated functional annotation, and biological function were identified using Glyma1.1, Glyma1.0, and NCBI RefSeq gene models in Soybase (www.soybase.org) with consideration for those candidate genes that may have a direct biological connection with canopy wilting, transpiration, rooting, or water transport.
Results
Phenotype descriptions
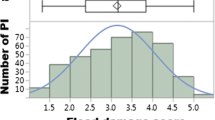
A broad range of canopy wilting within each environment indicated wide phenotypic variation. Canopy wilting scores had a range of 25 (PT-16), 25 (RO-16), 30 (SA-15), and 38 (SA-16) (Table 1). The Shapiro–Wilk test of normality was performed, which indicated that canopy wilting data were normally distributed within each environment and skewness and kurtosis also indicated a normal distribution (Table 1). Analysis of variance indicated that genotype, environment, and their interaction had significant effects (P ≤ 0.05) on canopy wilting. A significant positive correlation for canopy wilting between environments ranged from r = 0.40 between PT-16 and SA-15 to r = 0.66 between RO-16 and SA-16. Broad sense heritability of canopy wilting on an entry-mean basis was moderate to high for PT-16 (59%), RO-16 (74%), SA-15 (69%), and SA-16 (84%). When considering all environments, heritability was 80%.
Genomic best linear unbiased prediction (gBLUP) values were calculated utilizing the genomic-relationship matrix and phenotypic data of 373 accessions to estimate the genomic breeding values. The 373 accessions were ranked from lowest to highest based on the average gBLUP values of canopy wilting across all environments. Based on the average gBLUP-value ranking, the 15 accessions with lowest gBLUP for canopy wilting and 15 accessions with highest gBLUP for canopy wilting were selected (Table 2). Ranking of these 30 accessions was consistent with ranking of average phenotypic data of canopy wilting across all environments (Table 2). PI 592940 had the lowest canopy wilting for both gBLUP and phenotypic data, and PI507407 had the highest canopy wilting scores for both gBLUP and phenotypic data. These genotypes likely represent the most consistent extremes for canopy wilting.
Slow-wilting extreme PI 592940 had an average wilting score across environments of 11; in comparison, the slow-wilting check (PI 416937) had an average wilting score across environments of 20. Fast-wilting extreme PI 507407 had an average wilting score across environments of 39; in comparison, the fast-wilting check (A5959) had an average wilting score across environments of 33. The 15 accessions with lowest gBLUP values and canopy wilting scores averaged across all environments were from China (8 accessions), South Korea (6 accessions), and Taiwan (1 accession) (Table 2). The 15 accessions with the highest gBLUP values and canopy wilting scores averaged across all environments were from Japan (9 accessions), South Korea (5 accessions), and Georgia (1 accession). The breeding value and number of favorable alleles of these 30 accessions were calculated using allelic effects of significant SNPs. Slow-wilting accessions had large negative breeding values associated with reduced canopy wilting (−73 to −4). In contrast, fast-wilting accessions had large positive breeding values (38–106) associated with increased canopy wilting. Slow-wilting accessions had more favorable alleles (29–41) as compared to fast-canopy wilting accessions (10–24).
Markers distribution and linkage disequilibrium
Of the 31,260 markers used for association analysis, 22% of the markers had an MAF between 0.05 and 0.1, and 15% of the markers had a MAF between 0.10 and 0.15 (Table 3). Markers were more densely distributed in euchromatic (75.4%) than heterochromatic regions (24.6%) across the chromosomes. Out of a total of 950.1 Mb in the soybean genome, SNP density in euchromatic region ranged from 42 SNPs/Mb for Gm19 to 65 SNPs/Mb for Gm09. For the heterochromatic regions, SNP density ranged from 4 SNPs/Mb for Gm20 to 36 SNPs/Mb for Gm18. The average \( r^{2} \) between markers in the euchromatic region declined to 0.25 within 150 kb, whereas the r 2 in the heterochromatic region declined to 0.25 within 5000 kb (data not shown). These LD estimated results were consistent with results of Dhanapal et al. (2015b) and Hwang et al. (2014), indicating considerably greater LD for the euchromatic region than the heterochromatic region.
Genome-wide association analysis
The FarmCPU and MLM models were compared using the average canopy wilting data across all environments. The Q–Q plot of the FarmCPU model resulted in a sharp deviation from the expected P value distribution in the tail area, indicating that false-positives and negatives were adequately controlled, whereas Q–Q plots from MLM models did not show a sharp deviation (Fig. 1). These results indicated that FarmCPU was a better choice than MLM model for association testing in this study.

Quantile–quantile (QQ) plot of the mixed linear model (MLM) and fixed and random model circulating probability unification (FarmCPU) model using average canopy wilting data across all environments
Association analysis identified 61 significant SNPs in four environments associated with canopy wilting at the level of −Log10 (P) ≥ 3.5; P ≤ 0.0003 (Fig. 2). Out of 61 SNPs, 21 SNPs were present in at least two environments. One significant SNP on Gm20 was present in all four environments (Table 4). Significant SNPs, which were present within the same LD block, were considered as one locus, and out of the 61 significant SNPs identified across environments, there were 51 putative loci. Two putative loci on Gm06 and Gm16 were identified by three closely spaced SNPs, and five putative loci on Gm09, Gm13, Gm18, and Gm20 (2) were identified by two closely spaced SNPs, while the remaining loci were identified by one SNP. The allelic effect (difference in mean canopy wilting between genotypes with major allele and minor allele) for these significantly SNPs ranged from −7.40 to 5.18. A positive value indicates that the minor allele was the favorable allele associated with reduced canopy wilting and a negative value indicates that the major allele was the favorable allele associated with reduced canopy wilting. Information on the 61 significant SNPs, their corresponding MAF, major or minor allele, allelic effect, and common environments is listed in Table 4. These 61 significant SNPs from four environments were used to identify 61 genes within ± 10 kb of the respective SNPs. This distance was chosen, because it approximates the average distance between SNPs (18 kb). A list of these genes and their corresponding functional annotations (www.soybase.com) are provided (Table S1).

Manhattan plots of −Log10 (P) vs. chromosomal position of significant SNP associations and respective Quantile–quantile (QQ) plots of canopy wilting for four environments; a Salina 2015, b Salina 2016, c Pine Tree 2016, d Rohwer 2016, and e average canopy wilting across all environments (AAE) using the FarmCPU model. Red line represents the association threshold (−Log10 (P) ≥ 3.5; P ≤ 0.0003)
Association analysis of canopy wilting averaged across all environments identified 34 significant SNP associations at −Log10 (P) ≥ 3.5; P ≤ 0.0003 (Fig. 2). Of these 34 SNPs, seven were common to the 61 significant SNPs identified from four environments and among these seven SNPs, five SNPs were present in more than one environment (Table 5). Based on the closely spaced significant SNPs within the LD blocks, the 34 SNPs comprised 23 putative loci. The putative locus 1 on Gm01 was identified by 10 closely spaced SNPs, and locus 2 on Gm01 and locus 21 on Gm20 were identified by two closely spaced SNPs, while the remaining loci were all identified by one SNP. The allelic effect (difference in mean canopy wilting between genotypes with major allele and minor allele) for these significant SNPs ranged from −4.82 to 3.13. Information for these 34 significant SNPs, their corresponding MAF, major or minor allele, allelic effect, and common environments is listed in Table 5. These 34 significant SNPs from four environments were used to identify 34 genes within ±10 kb of the respective SNPs. A list of these genes and their corresponding functional annotations are provided (Table S2).
Discussion
This research evaluated canopy wilting in a panel of 373 MG IV soybean accessions in four environments along with slow-wilting (PI 416937) and fast-wilting (A5959) check genotypes. Canopy wilting had a wide range of phenotypic variation within each environment, which is important for dissecting complex traits through association mapping (McCarthy et al. 2008). In the panel of 373 accessions, genotypes were found with canopy wilting scores more extreme than any previous report. Two genotypes, PI 416937 and A5959, which were used in this study as checks, were also confirmed as slow- and fast-wilting in the previous research (Hwang et al. 2015, 2016; King et al. 2009). In the present research, PI 416937 and A5959 had average wilting scores across environments of 20 and 33, respectively (Fig. 3a). In comparison to the two checks, the average wilting score of slow-wilting PI 592940 was 11, and the average wilting score of the fast-wilting extreme PI 507407 was 39 (Table 2). Overall, 185 genotypes had lower average wilting scores across environments than did the slow-wilting check (PI 416937), but only two genotypes (PI507424 and PI507407) had wilting scores higher than the fast-wilting check. The genotypes with lower average wilting scores represent new genetic sources for the slow-wilting trait with potential alternative alleles or different mechanisms to achieve slow-wilting.

Distribution of average canopy wilting score across all environments (a), breeding values of accessions (b), and number of favorable alleles (c)
Carter et al. (2006) and King et al. (2009) reported that the accessions PI 416937 and PI 471938 were slow-wilting. The breeding values of these previously reported slow-wilting genotypes were 24 (PI 416937) and 20 (PI 471938) (Fig. 3b). In addition, the number of favorable alleles for these genotypes was 29 (PI 416937) and 24 (PI 471938) (Fig. 3c). In comparison, the most extreme slow-wilting accessions in this study had breeding values as low as -67 and as many as 41 favorable alleles (Table 2; Fig. 3b, c). The 15 accessions that were considered as slowest wilting in ranking also had considerably lower breeding values and more favorable alleles than PI 416937 and PI 471938. In contrast, the 15 accessions that were considered as fastest wilting in ranking had breeding values as high as 104 with only 10 favorable alleles. Hence, there is considerably greater variation in canopy wilting among genotypes than has been previously reported.
Significant positive correlations for canopy wilting between environments and a moderate-to-high heritability indicated that canopy wilting is a relatively stable trait across environments. Similar results of heritability were reported in several different mapping studies (Abdel-Haleem et al. 2012; Charlson et al. 2009; Hwang et al. 2015).
An advantage of GWAS over the traditional QTL mapping is that it is possible to map complex trait variation down to the nucleotide level. Out of 61 significant SNPs associated with canopy wilting, the minor alleles of 35 of these SNPs were favorable and associated with a decrease in the canopy wilting. One SNP with the minor allele associated with the largest reduction in the canopy wilting (5.18) was present on Gm18. This SNP was present within the coding region of Glyma18g14740, which encodes a protein functioning as a serine-glyoxylate amino-transaminase, and having a biological function involved with water transportation (www.soybase.com) (Table S1). Sinclair et al. (2008) concluded that low leaf hydraulic conductance in PI 416937 contributed to slow-wilting by conserving water under conditions of high evaporative demand. Interestingly, the SNP associated with the minor allele having the second largest reduction in canopy wilting was on Gm06 and was present in the coding region of Glyma06g45120. This region encodes the auxin-responsive GH3 family protein having a biological function associated with response of abscisic acid stimulus (Table S1). Biological functions of water transport and abscisic acid stimulus may directly result in phenotypic variation in canopy wilting. Presuming that slow-wilting is primarily associated with conserving soil moisture when soil moisture is plentiful (King et al. 2009; Ries et al. 2012) or by limiting transpiration when vapor pressure deficit is above a threshold value (Fletcher et al. 2007; Sadok and Sinclair 2009), slow-wilting genotypes expectantly would have lower water transport and greater expression of abscisic acid synthesis compared to fast-wilting genotypes. Up-regulation of Glyma18g14740 and Glyma06g45120 may, therefore, confer slow-wilting in plants under water deficit conditions. Similarly, other genes (Table S1) functioning in water transportation, abscisic acid stimulus, and root developmental, etc. may have roles in decreasing wilting by accessing more water (e.g., deeper rooting) or conserving water.
For 26 out of 61 SNPs associated with canopy wilting, the minor alleles of these SNPs were unfavorable and associated with an increase in canopy wilting (negative value of allelic effect indicates that minor allele was associated with an increase in canopy wilting in Table 4). One SNP on Gm08 had an allelic effect of −7.4, indicating that the major allele was favorable and associated with decreased canopy wilting. This SNP was present in the coding region of acyl-CoA synthetase gene, which functions in long-chain fatty acid metabolism. Table S1 provides information on 61 genes associated with identified SNPs that may have some association with canopy wilting, which can be directly or indirectly related to transpiration or water conservation for improving drought tolerance.
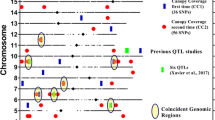
Several QTL mapping studies of canopy wilting have been conducted using different mapping populations to identify the chromosomal regions associated with canopy wilting variation. The studies found four QTLs (on chromosomes Gm08, Gm13, Gm14, and Gm17; Charlson et al. 2009), two QTLs (on chromosomes Gm08 and Gm20; Du et al. 2009), seven QTLs (on chromosomes Gm02, Gm04, Gm05, Gm12, Gm14, Gm17, and Gm19; Abdel-Haleem et al. 2012), and 22 QTLs (on chromosomes Gm02, Gm05, Gm11, Gm17, and Gm19; Hwang et al. 2015). Recently, meta-QTL analysis of canopy wilting by Hwang et al. (2016) reported nine meta-QTLs in eight chromosomal regions. Location of these reported chromosomal regions was compared with significant SNPs associated with canopy wilting in the present research from four environments and from the average across all environments (Fig. 4). Six different putative loci on five chromosomes Gm02 (2), Gm11, Gm17 (2), and Gm19 were located within six chromosomal regions that were identified by meta-QTL analysis of Hwang et al. (2016). These six putative loci consisted of eight significant SNPs, and six SNPs out of these eight had the minor allele associated with a decrease in the canopy wilting (Fig. 4).

Location of SNPs significantly associated with canopy wilting in four environments and across environments with previously identified QTLs for canopy wilting as shown in Soybase (www.soybase.org)
Some of the putative loci, located within previously reported chromosomal regions of meta-QTLs of canopy wilting (Hwang et al. 2016), may be candidates for decreasing wilting based on their biological functions. For example, Glyma02g01910, on Gm02, encodes a glycosyl hydrolase that has a biological function involved with root hair elongation. Up-regulation of Glyma02g01910 may increase root hair elongation in plants, providing more access to soil moisture and slowing wilting. A second gene, Glyma11g20020 on Gm11, encodes an acyl-CoA synthetase with a biological function involved in jasmonic acid biosynthesis. Jasmonic acid is an essential component of the signaling pathway in response to drought stress. Jasmonic acid is needed to induce ABA accumulation in roots of plants (de Ollas et al. 2015). Up-regulation of Glyma11g20020 may increase jasmonic acid biosynthesis, increasing ABA accumulation in roots, closing stomata, and conserving soil moisture.
Conclusions
In this study, high-density marker data of 31,260 SNPs with MAF ≥5% were used in GWAS to map the genomic regions controlling canopy wilting variation. Association analysis identified 61 significant SNPs associated with canopy wilting variation from four environments and 34 significant SNPs associated with average canopy wilting across all environments at a significance level of −Log10 (P) ≥ 3.5. Twenty-one significant associations of SNPs with canopy wilting out of 61 were present in at least two environments. The 61 SNP-canopy wilting associations and 34 SNPs identified from the average of canopy wilting across environments likely tagged 51 and 23 different loci, respectively. Six different putative loci were located within chromosomal regions that were previously reported as meta-QTLs for canopy wilting. Ultimately, significant SNPs that were present in more than one environment and those located within chromosome regions that were reported previously, are potential alleles for improving soybean drought tolerance. The genotypes identified with a large number of favorable slow-wilting alleles represent new genetic sources for crop improvement.
Author contribution statement
ASK and LCP designed the experiments, and ASK performed the statistical analysis. WTS, CAK, ASK, and LCP performed the field experiments and collected phenotypic data. ASK and JDR assembled the genotypic data. ASK co-wrote the manuscript with LCP. WTS, CAK, and JDR critically revised the manuscript. LCP coordinated and supervised the project. All authors read and approved the final manuscript.
References
Abdel-Haleem H, Carter TE Jr, Purcell LC, King CA, Ries LL, Chen PC, Schapaugh W Jr, Sinclair TR, Boerma HR (2012) Mapping of quantitative trait loci for canopy-wilting trait in soybean (Glycine max (L) Merr). Theor Appl Genet 125:837–846
Blum A (2005) Drought resistance, water-use efficiency, and yield potential-are they compatible, dissonant, or mutually exclusive? Aust J Agric Res 56:1159–1168
Blum A (2011) Drought resistance—is it really a complex trait? Funct Plant Biol 38(10):753–757
Bondari K (2003) Statistical analysis of genotype × environment interaction in agricultural research. In: Paper SD15, SESUG: The Proceedings of the SouthEast SAS Users Group, St Pete Beach
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Carter TE Jr, Orf JH, Purcell LC, Specht JE, Chen P, Sinclair T, Rufty TW (2006) Tough times, tough plants—new soybean genes defend against drought and other stresses. In: Proceedings of the 33rd Soybean Seed Research Conference, Chicago, IL, 5–8 Dec 2006
Carter TE Jr, De Souza PI, Purcell LC (1999) Recent advances in breeding for drought and aluminum resistance in soybean. In: Kauffman H (ed) Proceedings of the sixth World Soybean Research Conference, Chicago, IL. Superior Printing, Champaign, pp 106–125
Charlson DV, Bhatnagar S, King CA, Ray JD, Sneller CH, Carter TE Jr, Purcell LC (2009) Polygenic inheritance of canopy wilting in soybean [Glycine max (L) Merr]. Theor Appl Genet 119:587–594
Clark SA, van der Werf J (2013) Genomic best linear unbiased prediction (gblup) for the estimation of genomic breeding values. Genome-wide association studies and genomic prediction. Springer, Berlin, pp 321–330
de Ollas C, Arbona V, Gomez-Cadenas A (2015) Jasmonic acid interacts with abscisic acid to regulate plant responses to water stress conditions. Plant Signal Behav 10:e1078953. doi:10.1080/15592324.2015.1078953
Devi MJ, Sinclair TR (2013) Nitrogen fixation drought tolerance of the slow-wilting soybean PI 471938. Crop Sci 53:2072–2078
Dhanapal AP, Ray JD, Singh SK, Hoyos-Villegas V, Smith JR, Purcell LC, King CA, Fritsch FB (2015a) Association mapping of total carotenoids in diverse soybean genotypes based on leaf extracts and high-throughput canopy spectral reflectance measurements. PLoS One 10(9):e0137213
Dhanapal AP, Ray JD, Singh SK, Hoyos-Villegas V, Smith JR, Purcell LC, King CA, Cregan PB, Song Q, Fritsch FB (2015b) Genome-wide association study (GWAS) of carbon isotope ratio (δ13C) in diverse soybean [Glycine max (L.) Merr.] Genotypes. Theor Appl Genet 128:73–91
Dixit S, Huang BE, Sta Cruz MT, Maturan PT, Ontoy JCE, Kumar A (2014) QTLs for tolerance of drought and breeding for tolerance of abiotic and biotic stress: an integrated approach. PLoS One 9:e109574
Du W, Yu D, Fu S (2009) Detection of quantitative trait loci for yield and drought tolerance traits in soybean using a recombinant inbred line population. J Integr Plant Biol 51:868–878
Fletcher AL, Sinclair TR, Allen LH Jr (2007) Transpiration responses to vapor pressure deficit in well-watered ‘slow-wilting’ and commercial soybean. Environ Exp Bot 61:145–151
Fox CM, Cary TR, Colgrove AL, Nafziger ED, Haudenshiel JS, Hartman GL, Specht JE, Diers BW (2013) Estimating soybean genetic gain for yield in the northern United States-Influence of cropping history. Crop Sci 53:2473–2482
Hao D, Cheng H, Yin Z, Cui S, Zhang D, Wang H, Yu D (2012) Identification of single nucleotide polymorphisms and haplotypes associated with yield and yield components in soybean (Glycine max) landraces across multiple environments. Theor Appl Genet 124:447–458
Hill WG, Weir BS (1988) Variances and covariance of squared linkage disequilibria in finite populations. Theor Popul Biol 33:54–78
Hwang E, Song Q, Jia G, Specht JE, Hyten DL, Costa J, Cregan PB (2014) A genome-wide association study of seed protein and oil content in soybean. PLoS Genet 15:1
Hwang S, King CA, Chen P, Ray JD, Cregan PB, Carter TE Jr, Li Z, Abdel-Haleem H, Matson KW, Schapaugh W Jr, Purcell LC (2015) Confirmation of delayed canopy wilting QTLs from multiple soybean mapping populations. Theor Appl Genet 128:2047–2065
Hwang S, King CA, Chen P, Ray JD, Cregan PB, Carter TE Jr, Li Z, Abdel-Haleem H, Matson KW, Schapaugh W Jr, Purcell LC (2016) Meta-analysis to refine map position and reduce confidence intervals for delayed-canopy-wilting QTLs in soybean. Mol Breed 36:91
Irwin S, Good D (2015) Forming expectations for the 2015 U.S. Average soybean yield: what does history teach us? Farmdoc daily (5):51, Department of Agricultural and Consumer Economics, University of Illinois at Urbana-Champaign, March 19, 2015
King CA, Purcell LC, Brye KR (2009) Differential wilting among soybean genotypes in response to water deficit. Crop Sci 49:290–298
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ, Gore MA, Buckler ES, Zhang Z (2012) GAPIT: genome association and prediction integrated tool. Bioinformatics 28:2397–2399
Liu X, Huang M, Fan B, Buckler ES, Zhang Z (2016) Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet 12(2):e1005767
McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JPA, Hirschhorn JN (2008) Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nature Rev Genet 9(5):356–369
Money D, Gardner K, Migicovsky Z, Schwaninger H, Zhong GY, Myles S (2015) LinkImpute fast and accurate genotype imputation for non-model organisms. G3 5(11):2383–2390
Nordborg M, Borevitz J, Bergelsom J, Berry C, Chory J, Hagenbland J, Kreitman M, Maloof J, Noyes T, Oefner P, Stahl E, Weigel D (2002) The extent of linkage disequilibrium in Arabidopsis thaliana. Nat Genet 30(2):190–193
Oya T, Nepomuceno AL, Numaier N, Farias JRB, Tobita S, Ito S (2004) Drought tolerance characteristics of Brazilian cultivars—evaluation and characterization of drought tolerance of various Brazilian soybean cultivars in the field. Plant Prod Sci 7:129–137
Ray JD, Dhanapal AP, Singh SK, Hoyos-Villegas V, Smith JR, Purcell LC, King CA, Boykin D, Cregan PB, Song Q, Fritschi FB (2015) Genome-wide association study of ureide concentration in diverse maturity group IV soybean [Glycine max (L.) Merr.] accessions. G3 5(11):2391–2403
Ries LL, Purcell LC, Carter TE Jr, Edwards JT, King CA (2012) Physiological traits contributing to differential canopy wilting in soybean under drought. Crop Sci 52:272–281
Sadok W, Sinclair TR (2009) Genetic variability of transpiration response to vapor pressure deficit among soybean [Glycine max (L.) Merr.] cultivars. Crop Sci 49:955–960
SAS Institute (2013) The SAS System for Windows. Version 9.3. SAS Inst. Inc., Cary, NC
Sinclair TR, Zwieniecki MA, Holbrook NM (2008) Low leaf hydraulic conductance associated with drought tolerance in soybean. Physiol Plant 132:446–451
Sloane RJ, Patterson RP, Carter TE Jr (1990) Field drought tolerance of a soybean plant introduction. Crop Sci 30:118–123
Song Q, Hyten DL, Jia G, Quigley CV, Fickus EW, Nelson RL, Cregan PB (2013) Development and evaluation of SoySNP50K, a high-density genotyping array for soybean. PLoS One 8(1):e54985
Tuberosa R, Salvi S, Giuliani S, Sanguineti MC, Bellotti M, Conti S, Landi P (2007) Genome-wide approaches to investigate and improve maize response to drought. Crop Sci 47:120–141
USDA-FAS (2016) https://apps.fas.usda.gov/psdonline/circulars/production.pdf. Accessed 17 July 2017
Wen Z, Tan R, Yuan J, Bales C, Du W (2014) Genome-wide association mapping of quantitative resistance to sudden death syndrome in soybean. BMC Genom 15:809
Yu J, Pressoir G, Briggs WH, Vroh BI, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Zhang Z, Todhunter RJ, Buckler ES, Van Vleck LD (2007) Technical note: use of marker-based relationships with multiple-trait derivative-free restricted maximal likelihood. J Anim Sci 85:881–885
Zhang Z, Ersoz E, Lai CQ, Todhunter RJ, Tiwari HK, Gore MA, Bradbury PJ, Yu J, Arnett DK, Ordovas JM, Buckler ES (2010) Mixed linear model approach adapted for genome-wide association studies. Nat Genet 42:355–360
Zhang J, Song Q, Cregan PB, Nelson RL, Wang X, Wu J, Jiang GL (2015) Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genom 16:217
Zhu C, Gore MA, Buckler ES, Yu J (2008) Status and prospects of association mapping in plants. Plant Genom 1:5–20
Zipper SC, Qiu J, Kucharik CJ (2016) Drought effects on US maize and soybean production: spatiotemporal patterns and historical changes. Environ Res Lett 11:094021
Acknowledgements
The authors gratefully acknowledge partial funding of this research from the United Soybean Board. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture. The USDA is an equal opportunity provider and employer. Excellent technical support from Marilynn Davies, Jody Hedge, and Linda Martin is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Communicated by Volker Hahn.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kaler, A.S., Ray, J.D., Schapaugh, W.T. et al. Genome-wide association mapping of canopy wilting in diverse soybean genotypes. Theor Appl Genet 130, 2203–2217 (2017). https://doi.org/10.1007/s00122-017-2951-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-017-2951-z