
Abstract
Aims/hypothesis
Antagonism of the glucagon receptor (GCGR) represents a potential approach for treating diabetes. Cpd-A, a potent and selective GCGR antagonist (GRA) was studied in preclinical models to assess its effects on alpha cells.
Methods
Studies were conducted with Cpd-A to examine the effects on glucose-lowering efficacy, its effects in combination with a dipeptidyl peptidase-4 (DPP-4) inhibitor, and the extent and reversibility of alpha cell hypertrophy associated with GCGR antagonism in mouse models.
Results
Chronic treatment with Cpd-A resulted in effective and sustained glucose lowering in mouse models in which endogenous murine Gcgr was replaced with human GCGR (hGCGR). Treatment with Cpd-A also led to stable, moderate elevations in both glucagon and glucagon-like peptide 1 (GLP-1) levels, which were completely reversible and not associated with a hyperglycaemic overshoot following termination of treatment. When combined with a DPP-4 inhibitor, Cpd-A led to additional improvement of glycaemic control correlated with elevated active GLP-1 levels after glucose challenge. In contrast to Gcgr-knockout mice in which alpha cell hypertrophy was detected, chronic treatment with Cpd-A in obese hGCGR mice did not result in gross morphological changes in pancreatic tissue.
Conclusions/interpretation
A GRA lowered glucose effectively in diabetic models without significant alpha cell hypertrophy during or following chronic treatment. Treatment with a GRA may represent an effective approach for glycaemic control in patients with type 2 diabetes, which could be further enhanced when combined with DPP-4 inhibitors.
Similar content being viewed by others


Introduction
Glucagon, a key hormone in maintaining glucose homeostasis and preventing hypoglycaemia, [1] is secreted by pancreatic alpha cells during the fasting state and in between meals. Acting via the glucagon receptor (GCGR), which is predominantly localised in the liver, glucagon increases hepatic glycogenolysis and gluconeogenesis with subsequent elevation in hepatic glucose output. The actions elicited by glucagon are critical for maintaining euglycaemic states under normal physiological conditions. Conversely, hyperglucagonaemia is associated with hyperglycaemia and diabetes under pathophysiological conditions [2, 3]. Both animals and human patients with diabetes present with various degrees of hyperglucagonaemia [4, 5]. The state of chronic hyperglucagonaemia is correlated with excess hepatic glucose output and hyperglycaemia in diabetic patients [6]. Finally, experimental suppression of hyperglucagonaemia has been shown to correct postprandial hyperglycaemia in diabetic patients [7].
Given the role of glucagon in the development and maintenance of diabetes in both humans and animals, inhibition of the glucagon signalling pathway may represent a potential new approach for diabetes treatment [3, 7]. Somatostatin, an inhibitor of insulin and glucagon secretion, has been well documented to have hypoglycaemic effects [8–10]. Gcgr-knockout mice (Gcgr −/− mice) present with lower plasma glucose levels in the absence of overt hypoglycaemia [11, 12] and are resistant to hyperglycaemia under conditions of insulin deficiency [13]. The administration of glucagon-neutralising antibodies has been shown to be effective in reducing circulating levels of glucagon and hyperglycaemia in various animal models [14–16]. Furthermore, Gcgr antisense oligonucleotides (ASOs) have been shown to improve glucose metabolism in ob/ob mice [17, 18]. Studies have shown that acute treatment with non-peptidyl small-molecule GCGR antagonists (GRAs) blocked glucagon-induced glucose excursions in mice [19–22], dogs [23] and humans [24]. Our laboratory has recently reported [25, 26] that acute treatment with GRAs elicits similar effects on glucagon-induced glucose excursions in mice expressing the human GCGR (hGCGR) in place of the murine Gcgr. Finally, chronic treatment with a GRA was shown to result in sustained glucose lowering and improved pancreatic islet function in mice [27].
Previous reports showed Gcgr −/− mice possess a favourable metabolic phenotype, including normal glucose metabolism when fed a regular diet [11, 12]. In addition, Gcgr −/− mice are resistant to diet-induced obesity and streptozotocin (STZ)-mediated beta cell loss and hyperglycaemia [28]. However, Gcgr −/− mice have high circulating levels of glucagon and glucagon-like peptide 1 (GLP-1) accompanied by dramatic pancreatic alpha cell hypertrophy. Similar results have been seen in mice treated with Gcgr ASOs [17, 18]. These observations suggest that inhibition of glucagon signalling in Gcgr −/− and Gcgr ASO-treated mice leads to strong compensatory responses involving a marked alpha cell hypertrophy with associated upregulation of glucagon and GLP-1 production and/or secretion.
A critical question surrounding the use of small-molecule GRAs for the treatment of diabetes is whether or not these agents will elicit compensatory responses similar to those observed in Gcgr −/− and Gcgr ASO-treated mice. Potential treatment-induced elevations in glucagon and GLP-1 could influence the overall efficacy of GRA therapy. For competitive antagonists of GCGR, elevated levels of circulating glucagon may compete with GRAs for binding to GCGR, thereby reducing antagonism of the receptor as well as the overall therapeutic efficacy of this agent in vivo. In contrast, increased levels of GLP-1 may lead to significant improvements in glucose-dependent insulin secretion and postprandial glucose metabolism. Circulating GLP-1 levels elevated by GRA treatment also suggest potential for combination therapy with a GRA and a dipeptidyl peptidase-4 (DPP-4) inhibitor for enhanced the glycaemic efficacy. Depending on the magnitude and reversibility of a compensatory response to GCGR antagonism, the ensuing hyperglucagonaemic state may lead to transient or persistent hyperglycaemia when GRA therapy is terminated.
The current report describes a series of studies designed to examine the preclinical effects of treatment with a novel, small-molecule GRA (Cpd-A) on glucose-lowering as well as the extent, reversibility, metabolic consequences and mechanism of potential compensatory responses stemming from GRA treatment.
Methods
Animals
All animal procedures were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of Merck. Gcgr −/− mice [11] and hGCGR mice [25, 29] were generated and maintained as previously reported [30]. Unless otherwise noted, animals were fed standard rodent chow and water.
In vitro binding and functional assays
Investigations of the ability of compound A (Cpd-A) to bind to GCGR on isolated cell membrane and to inhibit glucagon-stimulated cAMP production in intact cells were performed as previously described [25]. For Schild analysis, the ability of glucagon to stimulate cAMP production in the intact Chinese hamster ovary (CHO)-hGCGR cells was measured in the presence of increasing concentrations of Cpd-A.
In vivo receptor occupancy assays
To determine the ability of Cpd-A to bind to glucagon receptors in liver in vivo, an in vivo receptor occupancy assay were performed in hGCGR mice as previously described [29]. Briefly, 1 h after Cpd-A administration, animals in the vehicle group were injected via the tail vein with either 125I-labelled glucagon alone or 125I-labelled glucagon plus excess unlabelled glucagon. Animals in Cpd-A group were treated with 125I-labelled glucagon alone in a similar fashion. Tissues were harvested 15 min after radiotracer injection, lysed and counted to determine levels of 125I-labelled glucagon in tissue.
Glucagon challenge assay
Glucagon challenge assays were performed as previously described [29]. Briefly, 1 h after Cpd-A administration, animals were challenged with either NaCl (vehicle group; negative control) or glucagon 15 μg/kg body weight (vehicle and Cpd-A groups) through i.p. injection. Blood glucose was measured via tail-vein bleeding using a glucometer (Lifescan, Milpitas, CA, USA) at the times indicated.
Chronic studies of obese hGCGR mice
Male hGCGR mice were fed a high-fat diet (HFD) (S3282: Bio-Serv, Frenchtown, NJ, USA) for 5 months to induce obesity. Obese hGCGR mice with blood glucose >7.5 mmol/l were selected for subsequent evaluation in which Cpd-A was administered as an admixture in the chow to reach the desired dose. Lean hGCGR mice maintained on normal chow were used as controls. In washout periods, animal were fed the same diet without Cpd-A. Pancreatic tissues were obtained at the end of the study for immunohistochemical staining. The oral glucose tolerance test (OGTT) was performed as previously described by Conarello et al. [28].
Efficacy studies in hGCGR HFD/STZ diabetic mice
HFD/STZ mice were prepared as previously described [31]. Briefly, hGCGR mice were fed an HFD (D12492; Research Diets, New Brunswick, NJ, USA) for 3 weeks before they were injected once (i.p.) with a low dose of STZ, 100 mg/kg (Sigma Chemicals, St Louis, MO, USA), to induce partial insulin deficiency. Three weeks after STZ injection, the majority of hGCGR HFD/STZ mice displayed hyperglycaemia, insulin resistance and glucose intolerance [31]. Cpd-A, des-fluoro-sitagliptin [31] (Merck Research Laboratories, Rahway, NJ, USA), and a combination of the two were dosed as an admixture to reach the desired dosage. Active glucagon-like peptide 1 (GLP-1) was measured using terminal (by cardiac puncture) plasma collected 5 min after a glucose challenge at 2 g/kg body weight.
Plasma hormone measurements
Glucagon and active GLP-1 were measured using commercial ELISA kits (Linco Research Immunoassay, St Charles, MO, USA). Inactive GLP-1 ELISA assay detecting GLP-19–37 was developed locally using an antibody specific to amino acid residues 9–37 of GLP-1, which detects both GLP-19–36 amide and GLP-19–37 but not other forms of GLP-1 [32]. Insulin was measured by ELISA with a commercial kit (ALPCO Diagnostics, Windham, NH, USA).
Immunohistochemical staining and analysis of pancreatic tissues
Immunolabelling was performed on pancreatic tissues as previously described [31]. Briefly, cryostat sections were immunostained with a combination of rabbit anti-glucagon antibody and guinea pig anti-insulin antibody. The insulin-positive beta cell-to-total islet area and glucagon-positive alpha cell-to-total islet area ratios were calculated from digitised images. Images of ten randomly chosen islets were captured from each section and were averaged to calculate ratios for each animal. Final results were represented by the mean and standard error of the ratios of four animals per treatment group.
Calculations
All data are expressed as means ± SD. Appropriate ANOVA analysis was conducted using GraphPad Prism software.
Results
Compound A (Cpd-A) is a potent and competitive antagonist of hGCGR in vitro
The structure of Cpd-A is illustrated in Fig. 1a. Cpd-A inhibited the binding of 125I-labelled glucagon in membranes prepared from Chinese hamster ovary (CHO)-hGCGR cells in a dose-dependent fashion with an IC50 of ~34 nmol/l (Fig. 1b) [26]. In vitro receptor signalling assays using intact CHO-hGCGR cells showed that Cpd-A is able to inhibit glucagon-stimulated cAMP production in a dose-dependent fashion with an IC50 of ~92 nmol/l (Fig. 1c) [26]. Cpd-A was similarly able to inhibit the murine receptor stably transfected in CHO cells with an IC50 of 398 nmol/l (data not shown). Schild analysis showed that Cpd-A inhibited glucagon-stimulated cAMP production in CHO-hGCGR cells with an equilibrium binding (association) constant (K B) of ~56.2 nmol/l (Fig. 1d). These collective results demonstrate that Cpd-A is a competitive inhibitor of glucagon binding to hGCGR and that the effects of this agent are approximately fourfold more potent for hGCGR than for murine GCGR. Further to this observed species specificity, all subsequent in vivo evaluations with Cpd-A were performed in mice expressing hGCGR rather than mouse-endogenous Gcgr [30].

In vitro properties of Cpd-A. a Structure of Cpd-A, 4-({9-tert-butyl-2-oxo-3-[4-(trifluoromethoxy)phenyl]-1,3-diazaspiro[5.5]undec-1-yl}methyl)-N-(2H-tetrazol-5-yl)benzamide. b Inhibition of glucagon binding on membranes. The results of a single representative study showing the binding of 125I-labelled glucagon to CHO-hGCGR cell membranes in the presence of increasing concentrations of Cpd-A. Data are shown as mean ± SD. c Inhibition of glucagon-stimulated cAMP production. The results of a single representative study showing the production of glucagon-stimulated cAMP in intact CHO-hGCGR cells in the presence of increasing concentrations of Cpd-A. Data are shown as mean ± SD. d Schild plot: titration of glucagon-stimulated cAMP production in intact CHO-hGCGR cells in the presence or absence of increasing concentrations of Cpd-A; K B = 56.2 nmol/l
Cpd-A binds and functionally antagonises hGCGR in vivo
Previous reports have shown that Cpd-A exhibits a favourable pharmacokinetic profile in mice with a half-life of 3.5 h and an oral bioavailability of 20% [26]. Therefore, receptor occupancy experiments were performed in hGCGR mice to determine whether Cpd-A binds to hepatic hGCGR in vivo. Approximately 75% of 125I-labelled glucagon present in liver occurred because of specific binding of the radiolabelled ligand to its receptor (Fig. 2a, lane 2 vs 1). Treatment with Cpd-A at 1, 3, and 10 mg/kg doses reduced the specific binding of 125I-labelled glucagon by 26%, 39% and 53%, respectively (Fig. 2b). These results demonstrate that Cpd-A binds to the hepatic hGCGR in vivo.

Cpd-A binds directly to hepatic hGCGR and blocks glucagon-induced glucose excursions in hGCGR mice. Levels of 125I-labelled glucagon in hepatic tissues (a) and the calculated percentage of hepatic hGCGR occupancy (b) from in vivo receptor-occupancy assays performed to assess the binding of Cpd-A to hepatic GCGR at 1 h after oral dosing. A glucagon challenge assay was performed to assess the ability of Cpd-A to block glucagon-induced glucose excursions. Blood glucose was measured at time points indicated in (c) and glucose AUC changes from time 0 were calculated (d). Significance was determined by ANOVA analysis; *p < 0.05; **p < 0.01; ***p < 0.001 vs glucagon-treated control. a–c show data as mean ± SEM. In c, white circles, vehicle; black squares, glucagon; white triangles, Cpd-A, 3 mg/kg; black triangles, Cpd-A, 10 mg/kg
The glucagon-challenged hGCGR mice showed a pronounced glucose excursion compared with vehicle-treated mice. Treatment with Cpd-A reduced the glucagon-induced glucose excursions (Fig. 2c, d), demonstrating that Cpd-A functionally antagonises GCGR in vivo.
Chronic treatment with Cpd-A in high-fat diet (HFD)-fed hGCGR mice lowers glucose and concomitantly raises circulating glucagon levels
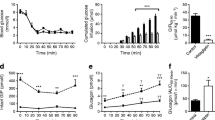
To evaluate whether chronic treatment with GRA improves glucose metabolism in vivo, obese hGCGR mice fed an HFD were dosed with Cpd-A at 1, 3 or 10 mg kg−1 day−1 (as admixture to the HFD) for 11 consecutive days. Food intake or body weight was not changed during the study (data not shown). An OGTT was conducted following dosing on day 11 using selected groups and the animals were killed (after blood glucose returned to pre-OGTT levels) to obtain plasma for measuring glucagon and inactive GLP-1 levels. Obese hGCGR mice had elevated plasma glucose levels compared with lean hGCGR mice fed a normal diet. Treatment of obese hGCGR mice with Cpd-A lowered plasma glucose levels in a dose-dependent fashion on days 3 and 11. Obese hGCGR mice treated with Cpd-A at 3 or 10 mg kg−1 day−1 showed similar plasma glucose levels compared with untreated, lean hGCGR mice on days 3 and 11 (Fig. 3a). The magnitude of the reduction in plasma glucose seen with Cpd-A at 3 and 10 mg kg−1 day−1 was similar on days 3 and 11. Furthermore, obese hGCGR mice treated with Cpd-A at 10 mg kg−1 day−1 showed an improvement in fasting glucose (4.7 ± 0.4, 3.8 ± 0.5 [p < 0.05 vs HFD control] and 4.3 ± 0.4 [p < 0.05 vs HFD control] mmol/l, respectively for HFD control, HFD Cpd-A and lean groups) as well as oral glucose tolerance on day 11 compared with that of untreated, obese hGCGR mice (Fig. 3b; baseline-subtracted AUC). These results demonstrate that Cpd-A produced sustained glucose lowering and improved glycaemic control throughout the course of chronic treatment.

Treatment with Cpd-A improves glucose metabolism with concomitant elevations in circulating glucagon levels in hGCGR mice. Obese hGCGR mice fed an HFD were treated with Cpd-A to achieve a dosage of 1, 3 or 10 mg kg−1 day−1 for a total of 11 days. The postprandial glucose levels at the beginning of the study (day 0, white bars; day 3, black bars; and day 11, hatched bars) following initiation of treatment are shown in (a); data are mean ± SD. In a similar study, an OGTT was performed on day 11 after overnight fasting using the Cpd-A 10 mg/kg group and the control group. A glucose load (2 g/kg) was administered at time 0 followed and blood glucose and insulin levels were measured at the time points indicated. Baseline (time 0)-subtracted glucose and insulin AUCs are shown in (b) and (c), respectively. White bars, HFD control; black bars, HFD Cpd-A 10 mg/kg; hatched bars, lean animal control. Circulating levels of glucagon obtained from terminal plasma samples are shown (mean ± SD) in (d). *p < 0.05; **p < 0.01 and ***p < 0.001 vs HFD control group, n = 8 in each group
Previous reports have shown that Gcgr −/− mice [12, 28] and mice treated with Gcgr ASOs [18] have a dramatic elevation in circulating levels of glucagon, suggesting that inhibition of the GCGR receptor results in a strong feedback or compensatory response on glucagon production and/or secretion. To determine whether treatment with a GRA produces a similar compensatory response, circulating levels of glucagon were measured in obese hGCGR mice following 11 day treatment with Cpd-A. Obese hGCGR mice treated with Cpd-A at 1, 3 or 10 mg kg−1 day−1 showed a dose-dependent increase in plasma glucagon of 1.3-, 2.0- and 2.7-fold relative to untreated obese hGCGR mice, respectively (Fig. 3d). These results demonstrate that pharmacological antagonism of the GCGR induces a compensatory response by raising glucagon levels; however, the extent of the hyperglucagonaemia elicited by Cpd-A was 2.7-fold at the highest dose tested compared with the 50- to 100-fold elevation in glucagon observed in the Gcgr −/− mice [11, 12]. These findings are consistent with a previous report showing that treatment with a GRA for 30 days resulted in an approximately threefold increase in circulating glucagon [27].
Enhanced glycaemic efficacy by treatment with Cpd-A in combination with des-fluoro-sitagliptin in hGCGR HFD/STZ mice
Des-fluoro-sitagliptin is a potent and selective DPP-4 inhibitor that has been shown to lower glucose levels and improve glycaemic control in a diabetic mouse model induced by a combination of HFD treatment and low-dose STZ injection [31]. The in vivo glycaemic effects of combination therapy with Cpd-A and des-fluoro-sitagliptin were evaluated in hGCGR HFD/STZ mice. Diabetic hGCGR HFD/STZ mice demonstrated marked hyperglycaemia compared with non-diabetic control mice as evidenced by an elevation in postprandial blood glucose levels (Fig. 4a). To examine the glycaemic efficacy of combination therapy, hGCGR HFD/STZ mice were treated with Cpd-A, des-fluoro-sitagliptin, or a combination of the two as admixtures to the HFD. At the end of the 2 week treatment, both Cpd-A and des-fluoro-sitagliptin monotherapy produced numerical reductions from baseline in blood glucose in hGCGR/STZ mice compared with untreated diabetic mice. In contrast, combination therapy with Cpd-A and des-fluoro-sitagliptin led to a significant reduction in blood glucose levels relative to untreated diabetic control mice (Fig. 4a).

Combination therapy with Cpd-A and a DPP-4 inhibitor led to improved glycaemic control. Diabetic hGCGR HFD/STZ mice were treated with Cpd-A (10 mg kg−1 day−1) or des-fluoro-sitagliptin (200 mg kg−1 day−1) alone or in combination for 2 weeks. Postprandial glucose (a) and OGTT were measured at day 14. OGTT was performed after 6 h fasting and glucose baseline (time 0)-subtracted AUC is shown in (b). Active GLP-1 (c) and insulin (d) levels were measured in plasma samples obtained at 5 min post glucose challenge using a subset of animals to detect peak GLP-1 levels and its effect on insulin secretion. White bars, diabetic control; black bars, Cpd-A; downward-hatched bars, des-fluoro-sitagliptin; grey bars, Cpd-A plus des-fluoro-sitagliptin; upward hatched bars, non-diabetic control. *p < 0.05; **p < 0.01; and ***p < 0.001 vs diabetic control group. † p < 0.001 vs combination group, n = 8–15 animals in each group
An OGTT study was performed in diabetic hGCGR HFD/STZ mice after 2 weeks of treatment to further evaluate the effects of combination therapy on glycaemic control. Animals were fasted for 6 h prior to OGTT and fasting glucose levels were 20.0 ± 1.0, 16.1 ± 1.1 (p < 0.05 vs diabetic control), 16.4 ± 1.4 (p < 0.05 vs diabetic control), 14.1 ± 1.4 (p < 0.01 vs diabetic control), and 8.4 ± 0.7 (p < 0.001 vs diabetic control) mmol/l, respectively, for diabetic control, Cpd-A, des-fluoro-sitagliptin, combination and non-diabetic control groups. Combination therapy resulted in significantly reduced glucose excursions as compared with diabetic control animals (Fig. 4b). Plasma samples were collected 5 min after the administration of a glucose challenge for the measurement of active GLP-1 levels and its effect on insulin secretion. Treatment with Cpd-A did not produce a significant effect on active GLP-1 levels compared with the diabetic control group. In contrast, treatment with des-fluoro-sitagliptin led to more than a twofold increase in active GLP-1 levels, which was further elevated (~ fivefold total vs diabetic control mice) by combination therapy (Fig. 4c), which correlates with increased insulin level (Fig. 4d). These findings suggest that combination treatment with a GRA agent and DPP-4 inhibitor produce synergistic effects on the biologically active form of GLP-1.
Prolonged treatment with Cpd-A leads to a moderate and stable elevation in glucagon without marked alpha cell hypertrophy in hGCGR mice
Gcgr−/− mice demonstrate alpha cell hypertrophy and dramatically elevated circulating levels of glucagon [12, 28]. To assess the extent of alpha cell hypertrophy following chronic treatment with GRAs, obese hGCGR mice were treated with Cpd-A at 10 mg kg−1 day−1 for up to 82 days. Consistent with results shown in Fig. 3d, treatment of obese hGCGR mice with Cpd-A significantly increased circulating levels of glucagon by 2.8-, 3.6- and 3.3-fold at days 11, 28, and 82, respectively relative to untreated obese hGCGR mice. Inactive GLP-1 levels were significantly increased by 2.2- and 1.7-fold relative to untreated obese hGCGR mice at days 28 and 82, respectively.
The potential effects of Cpd-A on pancreatic tissue were determined by immunohistochemical staining on pancreas sections prepared from obese hGCGR mice treated with Cpd-A at 10 mg kg−1 day−1 for 8 and 82 days (Fig. 5a–f). Pancreas from Gcgr −/− mice was included in the assay for comparison (Fig. 5b). Compared with lean hGCGR mice, pancreatic tissue from Gcgr −/− mice showed a ~4.6-fold increase in the ratio of glucagon-positive area to total area (G/T ratio). Obese hGCGR showed a ~50% lower G/T ratio relative to lean hGCGR mice (Fig. 5a, b, g). Chronic treatment with Cpd-A at 10 mg kg−1 day−1 increased the G/T ratio by approximately twofold in obese hGCGR mice compared with the ratio in vehicle-treated obese hGCGR mice and matched the G/T ratio observed in lean hGCGR mice at days 8 and 82. Compared with lean hGCGR mice, pancreatic tissues from Gcgr −/− mice showed a ~50% reduction in the ratio of insulin positive area to total area (I/T ratio). Untreated obese hGCGR mice demonstrated a mean increase of ~20% in I/T ratio relative to untreated lean hGCGR mice. Obese hGCGR mice chronically treated with Cpd-A at 10 mg kg−1 day−1 for 8 and 82 days showed a reduction in I/T ratio to levels that were comparable with those in lean hGCGR mice (Fig. 5a–f, h). These results demonstrate that prolonged treatment of obese hGCGR mice with Cpd-A does not cause overt alpha cell hypertrophy but rather results in moderate changes of alpha cell and beta cell mass to levels seen in lean hGCGR mice.

Prolonged treatment with Cpd-A does not induce alpha cell hypertrophy in hGCGR mice. Obese hGCGR mice fed an HFD were treated with Cpd-A at 10 mg kg−1 day−1. Immunohistochemical staining (a–f) was performed on pancreatic tissue samples collected at days 8 and 82 following treatment. Similar staining was also performed using pancreatic tissue samples obtained from lean hGCGR mice and Gcgr −/− mice. Green staining, insulin; red staining, glucagon. (a) Lean hGCGR; (b) lean Gcgr −/−; (c) HFD hGCGR day 8 control; (d) HFD hGCGR day 8 Cpd-A; (e) HFD hGCGR day 82 control; (f) HFD hGCGR day 82 Cpd-A. Ratios of areas staining positive for glucagon and insulin vs total area are shown in g, h. *p < 0.05 and ***p < 0.001 vs respective control groups (lean hGCGR for lean Gcgr −/− mice)
Cessation of Cpd-A treatment reverses hyperglucagonaemia without hyperglycaemia rebound
Studies were conducted to establish whether the mild glucagon elevation observed with Cpd-A treatment was reversible and whether rebound hyperglycaemia resulted upon termination of treatment. HFD-fed hGCGR mice were treated with Cpd-A at 10 mg kg−1 day−1 for 5 days and then the drug was withdrawn for 1 or 3 days. Plasma levels of glucagon and glucose were measured on days 1 and 3 of the washout period. The results showed that treatment of obese hGCGR mice with Cpd-A for 5 days significantly increased glucagon levels approximately twofold compared with control mice. Relative to day 5, plasma glucagon levels were corrected by ~50% or ~100% following 1 or 3 days of washout, respectively (Fig. 6a, b). The blood glucose levels in obese hGCGR mice following treatment with Cpd-A for 5 days were comparable with those in lean hGCGR mice. Relative to plasma glucose levels on day 5 of Cpd-A treatment, the removal of Cpd-A led to increases in plasma glucose of 17% and 14% on day 1 or 3 of washout, respectively. Nevertheless, the plasma glucose levels on days 1 and 3 following the termination of treatment were significantly lower than those seen in the HFD control group (Fig. 6c, d). Taken together, these results suggest that termination of Cpd-A treatment led to a rapid reversal of plasma glucagon elevation with no evidence of a hyperglycaemia overshoot.

Reversal of hyperglucagonaemia and lack of hyperglycaemia upon termination of Cpd-A treatment. hGCGR mice fed a HFD were treated with Cpd-A at 10 mg kg−1 day−1 for a total of 5 days. On day 5, the diet was switched to the regular HFD without Cpd-A. Plasma glucagon (a, b) and postprandial glucose (c, d) were measured on days 6 (24 h post treatment; a, c) and 8 (72 h post treatment; b, d). Data shown are mean ± SD. White bars, day 0; black bars, day 5; hatched bars, day 6 (i.e. 1 day washout); grey bars, day 8 (i.e. 3 day washout). *p < 0.05; **p < 0.01; and ***p < 0.001 vs indicated group
Discussion
Small-molecule GRAs represent a potential new therapeutic agent for diabetes treatment [7]. Acute treatment with a GRA has been shown to block glucagon-induced glucose excursions in various animal models [19–23, 25, 26, 29] and in humans [24]. In contrast, relatively little is known regarding the glucose-lowering efficacy of chronic GRA treatment in disease-relevant animal models. Studies from Gcgr −/− mice and mice treated with Gcgr ASOs suggest that inhibition of glucagon signalling could lead to compensatory increase of circulating glucagon and GLP-1, possibly as a result of alpha cell hypertrophy [28]. It is not known whether pharmaceutical inhibition of GCGR will elicit similar compensatory responses and, more importantly, how elevated levels of glucagon and GLP-1 might affect GRA chronic efficacy. We report here that Cpd-A binds to hepatic GCGR and functionally antagonises GCGR action when administered acutely in vivo. Treatment with Cpd-A for 11 days effectively improved glycaemic control in HFD-fed hGCGR mice, demonstrating that chronic treatment with GRA sustained glucose lowering in mice with diet-induced obesity. The results of the current studies demonstrate that treatment with Cpd-A increases circulating levels of glucagon and GLP-1 moderately (i.e. two- to threefold elevation vs vehicle) relative to those observed in Gcgr −/− and Gcgr ASO treated mice. Similar levels of glucagon and GLP-1 were seen following 11, 28 and 82 days of treatment with Cpd-A, suggesting that the compensatory response to GRA treatment stabilised within the first 11 days of treatment and did not continue to increase over time. This agrees well with a previous report that chronic treatment with a structurally distinct GRA for 30 days resulted in an approximately threefold increase in plasma glucagon levels [27].
In contrast to dramatic changes in islet cell morphology and significant alpha cell hypertrophy in Gcgr −/− mice observed in previous studies [11, 12, 28], chronic treatment with Cpd-A for up to 82 days did not lead to detectable changes in the islet cell morphology. A moderate increase or decrease in the area of pancreatic tissue stained positive for glucagon or insulin, respectively, was observed when compared with HFD-treated vehicle control mice. These pancreatic changes induced by Cpd-A actually made the tissue closer to that of healthy lean hGCGR mice, suggesting that chronic GRA therapy reversed the abnormalities seen in untreated obese hGCGR mice. These results indicate that chronic GRA treatment does not induce alpha cell hypertrophy and may improve pancreatic function as suggested by Winzell et al. [27]. So, data obtained from these studies demonstrate that chronic GRA treatment using a compound such as Cpd-A represents a potential means of effectively maintaining glucose lowering and is unlikely to induce adverse events such as overt hyperglucagonaemia or alpha cell hypertrophy. While feasible, it may also be critical to maintain a balance of potency and safety profile for therapy of this mechanism. This becomes more important when considering the relatively short treatment period of our studies vs the lifetime GCGR blockage in Gcgr −/− mice. It also cannot be ruled out completely that lack of alpha cell hypertrophy with Cpd-A is partially due to its relatively moderate potency, even though alpha cell hypertrophy was not observed with some significantly more potent molecules tested in a similar paradigm (data not shown).
The mechanism behind GRA-mediated mild glucagon and GLP-1 elevations, in contrast to much greater increases in Gcgr −/− or Gcgr ASO-treated mice, is unclear. Partial receptor coverage as indicated by receptor occupancy results combined with changes in drug level over time could be part of the answer. Results from current studies suggest full receptor occupancy is not required for the biological action of GRA. It will be interesting to find out whether a more potent compound and longer treatment can cause further elevation of glucagon/GLP-1 levels. Elevated GLP-1 levels may contribute to the benefit seen with incomplete GCGR blockade alone. Additional potential players, e.g. leptin [13] and fibroblast growth factor 21 (FGF21), cannot be excluded and will be the subject of future studies. As results from washout studies clearly indicate, GRA-induced hyperglucagonaemia is reversible and is unlikely to cause hyperglycaemia following the termination of treatment. In contrast to concerns regarding glucagon elevation, increased GLP-1 in circulation (primarily the inactive form of GLP-1; data not shown) created a potential opportunity to combine a GRA with a DPP-4 inhibitor to achieve a higher level of active GLP-1 in the circulation and thus improve efficacy beyond that of individual therapy. Indeed, studies performed in hGCGR HFD/STZ mice show that combination treatment of Cpd-A with des-fluoro-sitagliptin led to enhanced glycaemic efficacy. Even though absolute improvement of glycaemic control by Cpd-A or des-fluoro-sitagliptin does not seem to be large or strictly additive in the severely diabetic HFD/STZ model, a clear increase in active GLP-1 and its effects on insulin secretion after a glucose challenge provided more evidence of the mechanism of action and the potential of the combination therapy. These results suggest that combination therapy with GRAs and DPP-4 inhibitors may have added benefit in treating diabetes when compared with individual therapy alone.
In summary, the current study demonstrated that Cpd-A functions as a potent and competitive antagonist of hGCGR both in vitro and in vivo. In mice, chronic treatment with Cpd-A resulted in sustained glucose lowering coincident with stable and moderate elevations in glucagon and GLP-1. Elevation of these hormones was reversible and did not result in a glucose overshoot upon termination of treatment. Furthermore, chronic treatment with Cpd-A was not associated with overt changes in the morphology of pancreatic tissues. These observations suggest that GRAs may represent an effective and safe therapy for improving glucose metabolism in humans. Furthermore, combination therapy with a GRA and a DPP-4 inhibitor could lead to enhanced glycaemic efficacy by combining the two mechanisms.
Abbreviations
- ASO:
-
Antisense oligonucleotides
- CHO:
-
Chinese hamster ovary
- Cpd-A:
-
Compound A
- DPP-4:
-
Dipeptidyl peptidase-4
- (h)GCGR:
-
(Human) glucagon receptor
- GLP-1:
-
Glucagon-like peptide-1
- GRA:
-
Glucagon receptor antagonist
- G/T:
-
Ratio of glucagon positive area to total area
- HFD:
-
High-fat diet
- I/T:
-
Ratio of insulin positive area to total area
- K B :
-
Equilibrium binding (association) constant
- STZ:
-
Streptozotocin
References
Unger RH (1971) Glucagon physiology and pathophysiology. N Engl J Med 285:443–449
Fanelli CG, Porcellati F, Rossetti P, Bolli GB (2006) Glucagon: the effects of its excess and deficiency on insulin action. Nutr Metab Cardiovasc Dis 16(Suppl 1):S28–S34
Jiang G, Zhang BB (2003) Glucagon and regulation of glucose metabolism. Am J Physiol Endocrinol Metab 284:E671–E678
Burcelin R, Katz EB, Charron MJ (1996) Molecular and cellular aspects of the glucagon receptor: role in diabetes and metabolism. Diabetes Metab 22:373–396
Toft I, Gerich JE, Jenssen T (2002) Autoregulation of endogenous glucose production during hyperglucagonemia. Metabolism 51:1128–1134
Consoli A (1992) Role of liver in pathophysiology of NIDDM. Diabetes Care 15:430–441
Zhang BB, de Moller (2000) New approaches in the treatment of type 2 diabetes. Curr Opin Chem Biol 4:461–467
Gerich JE, Lorenzi M, Schneider V et al (1974) Effects of somatostatin on plasma glucose and glucagon levels in human diabetes mellitus. Pathophysiologic and therapeutic implications. N Engl J Med 291:544–547
Raskin P, Unger RH (1978) Hyperglucagonemia and its suppression. Importance in the metabolic control of diabetes. N Engl J Med 299:433–436
Sakurai H, Dobbs R, Unger RH (1974) Somatostatin-induced changes in insulin and glucagon secretion in normal and diabetic dogs. J Clin Invest 54:1395–1402
Gelling RW, Du XQ, Dichmann DS et al (2003) Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci U S A 100:1438–1443
Parker JC, Andrews KM, Allen MR, Stock JL, McNeish JD (2002) Glycemic control in mice with targeted disruption of the glucagon receptor gene. Biochem Biophys Res Commun 290:839–843
Wang MY, Chen L, Clark GO et al (2010) Leptin therapy in insulin-deficient type I diabetes. Proc Natl Acad Sci USA 107:4813–4819
Brand CL, Rolin B, Jorgensen PN, Svendsen I, Kristensen JS, Holst JJ (1994) Immunoneutralization of endogenous glucagon with monoclonal glucagon antibody normalizes hyperglycaemia in moderately streptozotocin-diabetic rats. Diabetologia 37:985–993
Brand CL, Jorgensen PN, Knigge U et al (1995) Role of glucagon in maintenance of euglycemia in fed and fasted rats. Am J Physiol 269:E469–E477
Brand CL, Jorgensen PN, Svendsen I, Holst JJ (1996) Evidence for a major role for glucagon in regulation of plasma glucose in conscious, nondiabetic, and alloxan-induced diabetic rabbits. Diabetes 45:1076–1083
Liang Y, Osborne MC, Monia BP et al (2004) Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes 53:410–417
Sloop KW, Cao JX, Siesky AM et al (2004) Hepatic and glucagon-like peptide-1-mediated reversal of diabetes by glucagon receptor antisense oligonucleotide inhibitors. J Clin Invest 113:1571–1581
Lau J, Behrens C, Sidelmann UG et al (2007) New beta-alanine derivatives are orally available glucagon receptor antagonists. J Med Chem 50:113–128
Ling A, Hong Y, Gonzalez J et al (2001) Identification of alkylidene hydrazides as glucagon receptor antagonists. J Med Chem 44:3141–3149
Ling A, Plewe M, Gonzalez J et al (2002) Human glucagon receptor antagonists based on alkylidene hydrazides. Bioorg Med Chem Lett 12:663–666
Madsen P, Ling A, Plewe M et al (2002) Optimization of alkylidene hydrazide based human glucagon receptor antagonists. Discovery of the highly potent and orally available 3-cyano-4-hydroxybenzoic acid [1-(2,3,5,6-tetramethylbenzyl)-1H-indol-4-ylmethylene]hydrazide. J Med Chem 45:5755–5775
Rivera N, Everett-Grueter CA, Edgerton DS et al (2007) A novel glucagon receptor antagonist, NNC 25-0926, blunts hepatic glucose production in the conscious dog. J Pharmacol Exp Ther 321:743–752
Petersen KF, Sullivan JT (2001) Effects of a novel glucagon receptor antagonist (Bay 27-9955) on glucagon-stimulated glucose production in humans. Diabetologia 44:2018–2024
Qureshi SA, Rios CM, Xie D et al (2004) A novel glucagon receptor antagonist inhibits glucagon-mediated biological effects. Diabetes 53:3267–3273
Shen DM, Zhang F, Brady EJ et al (2005) Discovery of novel, potent, and orally active spiro-urea human glucagon receptor antagonists. Bioorg Med Chem Lett 15:4564–4569
Winzell MS, Brand CL, Wierup N et al (2007) Glucagon receptor antagonism improves islet function in mice with insulin resistance induced by a high-fat diet. Diabetologia 50:1453–1462
Conarello SL, Jiang G, Mu J et al (2007) Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia 50:142–150
Dallas-Yang Q, Shen X, Strowski M et al (2004) Hepatic glucagon receptor binding and glucose-lowering in vivo by peptidyl and non-peptidyl glucagon receptor antagonists. Eur J Pharmacol 501:225–234
Shiao LL, Cascieri MA, Trumbauer M, Chen H, Sullivan KA (1999) Generation of mice expressing the human glucagon receptor with a direct replacement vector. Transgenic Res 8:295–302
Mu J, Woods J, Zhou YP et al (2006) Chronic inhibition of dipeptidyl peptidase-4 with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes 55:1695–1704
Migoya EM, Bergeron R, Miller JL et al (2010) Dipeptidyl peptidase-4 inhibitors administered in combination with metformin result in an additive increase in the plasma concentration of active GLP-1. Clin Pharmacol Ther 88:801–808
Acknowledgements
We dedicate this work to the memory of our dearest colleague, R. Saperstein, who made a tremendous contribution to the investigation of the in vivo effect of glucagon antagonism. The authors wish to thank S. Lin for guiding compound synthesis, and M. Wu and C. Canasto-Chibuque (Merck) for their excellent technical support. The authors also wish to thank A. O. Johnson-Levonas and K. Newcomb (Merck) for their assistance in the editing and formatting this manuscript for publication. Financial support for this study was provided by Merck.
J.M. participated in the planning and design of the study, interpreted results, and wrote sections of the manuscript. G.J. and E.B participated in the planning and design of the study, collection of data, analysis and interpretation of results, and wrote sections of the manuscript. F.L. participated in the collection and analysis of data and review of the manuscript. B.B.Z. and J.W. participated in the collection and analysis of data, interpretation of results and review of the manuscript. E.Z. and R.S. participated in the collection, analysis and interpretation of data and review of the manuscript. M.W. participated in the planning and design of the study, collection and analysis of data. Z.L. participated in the planning and design of the study, collection and analysis of data and review of the manuscript. K.L. and Q.D-Y. participated in the planning and design of the study, collection and analysis of data, interpretation of results and review of the manuscript. L.Z. and X.S. participated in the collection and analysis of data and review of the manuscript. M.L.C. participated in the analysis of data, interpretation of results and review of the manuscript. S.A.Q. and D.M-S. participated in the interpretation of results and review of the manuscript. F.Z. participated in the planning and design of the study and review of the manuscript. E.R.P. participated in the planning and design of the study, interpretation of results and review of the manuscript. All authors have approved the final version of the manuscript.
Duality of interest
All authors are currently, or were, employees of Merck and may own stock or hold stock options in Merck.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. Mu, G. Jiang and E. Brady contributed equally to this study.
Rights and permissions
About this article
Cite this article
Mu, J., Jiang, G., Brady, E. et al. Chronic treatment with a glucagon receptor antagonist lowers glucose and moderately raises circulating glucagon and glucagon-like peptide 1 without severe alpha cell hypertrophy in diet-induced obese mice. Diabetologia 54, 2381–2391 (2011). https://doi.org/10.1007/s00125-011-2217-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2217-2