
Abstract
Type 2 diabetes prevalence is increasing dramatically across the globe, imposing a tremendous toll on individuals and healthcare systems. Reversing these trends requires comprehensive approaches to address both classical and emerging diabetes risk factors. Recently, environmental toxicants acting as endocrine-disrupting chemicals (EDCs) have emerged as novel metabolic disease risk factors. EDCs implicated in diabetes pathogenesis include various inorganic and organic molecules of both natural and synthetic origin, including arsenic, bisphenol A, phthalates, polychlorinated biphenyls and organochlorine pesticides. Indeed, evidence implicates EDC exposures across the lifespan in metabolic dysfunction; moreover, specific developmental windows exhibit enhanced sensitivity to EDC-induced metabolic disruption, with potential impacts across generations. Importantly, differential exposures to diabetogenic EDCs likely also contribute to racial/ethnic and economic disparities. Despite these emerging links, clinical practice guidelines fail to address this underappreciated diabetes risk factor. Comprehensive approaches to stem the tide of diabetes must include efforts to address its environmental drivers.
Similar content being viewed by others


Introduction
The global burden of diabetes is projected to reach a staggering 629 million individuals by 2045 [1], and the WHO envisages metabolic disorders will be a major cause of death by 2030, placing substantial economic stresses on healthcare systems worldwide [2]. Indeed, 12% of global health expenditures are already estimated to be spent on diabetes [1] and, in the USA alone, annual diabetes-associated economic costs are calculated to be $327 billion [3]. Most of the diabetes health burden arises from type 2 diabetes, which accounts for 90–95% of all diabetes cases. As both disease prevalence and treatment costs continue to rise dramatically [4], comprehensive approaches to reverse these trends are desperately needed. Essential to this effort are strategies that simultaneously address the myriad risk factors contributing to diabetes pathogenesis. While this certainly includes efforts to tackle the contributions of excess food intake and sedentary lifestyles, it is also essential to identify other modifiable risk factors that promote diabetes development.
One emerging but underappreciated realm of risk is our ubiquitous exposure to environmental chemicals through food and water, skin contact and the air we breathe. Indeed, exposures to endocrine-disrupting chemicals (EDCs) are associated with an increased risk of various endocrine-related disorders in both human and animal studies [5]. The Endocrine Society classifies exogenous chemicals as EDCs based upon their ability to disrupt any aspect of hormone secretion or action [6]. Over the last two decades, exposure to many EDCs from multiple sources has been associated with increased risk of diabetes, obesity and other metabolic disorders (Fig. 1). These metabolism-disrupting EDCs include a variety of inorganic and organic molecules of natural or synthetic origin, including many to which humans are frequently exposed, including arsenic, bisphenol A (BPA) and phthalates, as well as legacy compounds, which have profound chemical and biological stability and persist in the environment despite regulatory action to eliminate or curtail their use, such as polychlorinated biphenyls (PCBs) and organochlorine pesticides. With increasing evidence linking exposure to various EDCs with diabetes in human populations, as well as diabetes-associated pathophysiological defects in cell-based and animal studies [7,8,9,10] (Fig. 2), the time has come to incorporate environmental interventions into comprehensive strategies to reduce the individual and societal burden of this devastating disease.

Sources of EDCs. A diverse array of chemicals from various sources have been linked to metabolic dysfunction in cell-based, animal and epidemiological studies. These metabolism-disrupting EDCs include both inorganic and organic compounds of both natural and synthetic origin. Humans are exposed through the use, production and environmental dissemination of these chemicals in food production, industrial activity, and home and personal care, as well as through medical care. This figure is available as part of a downloadable slideset

EDCs associated with type 2 diabetes pathogenesis and their proposed mechanisms of action. Type 2 diabetes arises from a combination of reduced insulin sensitivity (solid black arrow) and progressive beta cell failure (dashed black arrow). The blue circles, labelled a to e, represent individual states. Starting at state ‘a’, (in the middle of the normoglycaemia curve, green shading), if a person with these levels of insulin sensitivity and insulin secretion were to exercise, lose weight and get better sleep, they would slide down the curve to ‘b’. Unfortunately, our society is instead less active, consuming excess food and sleeping less, driving individuals from state ‘a’ to state ‘c’. This situation is, however, untenable in the long run. As one’s beta cells begin to fail, they fall off the curve (dashed line) to ‘d’ (impaired glucose tolerance, yellow shading); this condition then further deteriorates to ‘e’ (type 2 diabetes, red). Several EDCs have been linked to altered insulin sensitivity (darker red text box), beta cell disruptions (darker blue text box), or both. Multiple mechanisms have been ascribed to EDC-induced beta cell dysfunction and altered functioning of insulin-responsive tissues (lighter blue and lighter red text boxes). Data supporting EDC-mediated diabetes pathogenesis are derived from: cell-, islet- and tissue-based studies; animal models; and epidemiological and clinical studies (indicated by the superscript numbers; purple text box). BPA, bisphenol A; DDE, dichlorodiphenyldichloroethylene; DDT, dichlorodiphenyltrichloroethane; GR, glucocorticoid receptor; OC, organochlorine; PBDEs, polybrominated diphenyl ethers; PCBs, polychlorinated biphenyls; PFASs, per- and polyfluoroalkyl substances; PCDD/Fs, polychlorinated dibenzo-p-dioxins and furans; PM, particulate matter; POPs, persistent organic pollutants; PPARs, peroxisome proliferator-activated receptors; TCDD, 2,3,7,8-tetrachlorodibenzodioxin. Data compiled from [7,8,9,10]. Figure adapted by permission from Springer Nature [129], ©2006 Nature Publishing Group. This figure is available as part of a downloadable slideset
Exposures during sensitive developmental windows
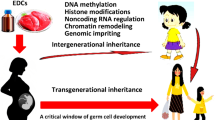
While EDC exposures occur across the lifespan, specific developmental windows are recognised as periods of unique vulnerability (Fig. 3). The period from conception to birth is a time of cellular replication and differentiation, functional organ system maturation and rapid growth. These processes are exquisitely sensitive to perturbations in the intrauterine metabolic milieu that exert long-lasting effects on offspring. Animal and epidemiology data demonstrate that several critical periods during development influence the later-life onset of type 2 diabetes, including preconception and gestation, early infancy, the adiposity rebound period between 5 and 7 years of age, and puberty. Until recently, most studies examining developmental programming were focused on inadequate nutrition or uteroplacental insufficiency; however, it is now apparent that developmental exposure to EDCs may also increase the incidence of type 2 diabetes [7, 11].

Influence of EDCs across the lifespan. EDCs and other environmental toxicants are an important component of the overall environmental milieu (blue) that interacts with genetic susceptibility to influence diabetes pathogenesis across the lifespan (green arrow). Importantly, exposures during sensitive windows can disrupt developmental programming and result in long-term metabolic dysfunction in both the exposed individual as well as future generations (black arrows). This figure is available as part of a downloadable slideset
Perhaps the prototypical EDC is BPA, a common contaminant used widely in polycarbonate and other plastics, epoxy resins lining cans and the thermal paper of cash register receipts. BPA binds both oestrogen receptor α (ERα) and β (ERβ), although with less affinity than 17β-oestradiol (E2). Humans are ubiquitously exposed to BPA, with low levels detectable in the blood and urine of virtually all individuals across multiple populations [12,13,14]. Importantly, human fetuses and children are likely to be at heightened risk of endocrine disruption from BPA, since BPA clearance enzymes are not fully functional at these life stages [15]. Consequently, blood and urine levels of BPA are higher in fetuses than in their pregnant mothers [13], and in infants and children compared with adults [12]. Importantly, while exact human exposures are difficult to estimate, animal studies demonstrate that levels below those considered safe for human health are sufficient to induce metabolic dysfunction [16].
Multiple animal studies have explored the impact of developmental BPA exposure on metabolic programming; however, controversy persists regarding the effects of maternal BPA exposure on offspring. Differences across studies, include species and strain, developmental stage during exposure, ages at metabolic interrogation, levels of exposure, housing, diet, control conditions and exposure routes have all contributed to a lack of consensus on the gestational effects of BPA. Particularly robust debate has focused upon the importance of the route of exposure (oral vs subcutaneous) as well as evidence of non-monotonic dose–response relationships demonstrating that BPA exposures at or below the current US Environmental Protection Agency’s reference dose often yield metabolic disruptions that are either not observed or different at higher doses [17,18,19]. Acknowledging these challenges, when experimental conditions are strictly maintained, reproducible and stable metabolic disruptions are consistently observed in the offspring of BPA-exposed dams [20].
Some of the first evidence that developmental EDC exposure alters metabolic health came from studies exposing pregnant rats to BPA. Exposure resulted in dose- and sex-specific increases in offspring body weights that persisted into adulthood [21]. Since then, multiple studies have examined links between perinatal EDC exposure and the later-life development of metabolic dysfunction, including obesity. Fewer studies have examined the effects of EDCs on beta cell development and function. Importantly, all three oestrogen receptors (ERα, ERβ and the G-protein coupled ER [GPER]) are present in rodent and human beta cells, where they play essential roles in islet survival and function [22]; thus, beta cells are primed for disruption by ER-active EDCs like BPA. Indeed, fetal offspring of C57BL/6 dams fed a BPA-containing diet from embryonic day 7.5 (E7.5) to E18.5 exhibited altered endocrine pancreas development, with disruptions in the number and relative cellular composition of islets [23]. Using similar exposures, male offspring of pregnant mice exposed subcutaneously to BPA exhibited enhanced insulin secretion at 17 weeks of age, suggesting BPA-induced insulin resistance; moreover, by 28 weeks, the mice exhibited hyperglycaemia without alterations in glucose-stimulated insulin secretion (GSIS) [24]. Of note, follow-up studies using a similar exposure window and a higher BPA dose demonstrated that the lower BPA dose, but not the higher dose, decreased GSIS at postnatal day 30, despite increased beta cell mass, which was attributed to increased expression of pro-proliferative genes and decreased expression of genes regulating beta cell death [25]. Intriguingly, beta cell mass was actually decreased in BPA-treated animals by postnatal day 120. The discrepancies in the two studies are likely to be related to the different postnatal ages examined; however, it remains unclear why GSIS was impaired in young animals but was normal at later ages.
To better mimic exposures during human development, we examined the impact of dietary exposure of mice to environmentally relevant BPA doses from 2 weeks prior to pregnancy to the end of lactation using a low phytoestrogen control diet and BPA-free cages; serum BPA levels approximated those in humans [17]. Male offspring of exposed dams, but not females, developed abnormal glucose tolerance associated with insulin secretory defects, including increased basal rates of insulin release at the higher BPA dose that was suggestive of insulin resistance. Interestingly, differences in insulin release using a glucose gradient were not observed between control males and those that received a higher dose of BPA, nor was KCl-induced insulin release in islets altered by the higher BPA dose, suggesting an intact insulin secretory apparatus. In contrast, male offspring exposed to the lower BPA dose exhibited similar basal rates of insulin release; however, maximal GSIS was impaired [17]. These data suggest that the metabolic consequences of developmental BPA exposure exhibit complex dose–response relationships.
An area of intense interest in the developmental programming of metabolism is the multigenerational heritability of metabolic traits after ancestral EDC exposure. To determine whether BPA-induced metabolic disruptions persisted into F2 offspring, BPA-exposed F1 females were mated to unexposed males [17]. Like F1 offspring, F2 males of dams exposed to a higher dose of BPA were glucose intolerant with preserved GSIS, while F2 males of dams exposed to a lower dose of BPA exhibited blunted GSIS. In addition, lower dose BPA exposure significantly reduced beta cell mass and increased beta cell death in F1 males, which persisted into the F2 generation; transcriptomic analyses indicated significant dose-specific changes in genes regulating inflammation and mitochondrial function [18]. Collectively, these results demonstrate multigenerational dose- and sex-specific metabolic effects of developmental BPA exposure.
These data fit within a broadening context of multigenerational and transgenerational effects of multiple EDC classes on various endocrine and metabolic tissues [7, 26,27,28,29]. Recently, studies have demonstrated that changes in the expression and methylation of imprinted genes in the brain persist across three generations after BPA exposure in mice [30]. In examining the transgenerational (F3) effects of BPA on metabolic health, we found that F3 adult males exposed ancestrally to either the lower or higher BPA dose had increased body weight with preserved glucose tolerance [19]. F3 male offspring of dams exposed to the lower BPA dose had reduced beta cell mass and smaller islets that exhibited enhanced GSIS. There was no effect of BPA in F3 females. These studies show that maternal BPA exposure resulted in fewer metabolic defects in F3 than F1 and F2 offspring, and these were sex- and dose-specific. Interestingly, while these data suggest a decay in EDC effects over generations, the relationships may be more complex for other exposures. For example, the fungicide and booster biocide tributyltin (TBT) has been shown to exert transgenerational effects on adipose tissue, with some phenotypes that amplify over successive generations [31].
Mechanisms of EDC action
There are a number of possible mechanisms by which EDCs can influence health outcomes. These include interference with the activity of nuclear receptors, noncanonical steroid hormone receptors and orphan receptors, as well as disruptions in the enzymatic pathways regulating steroid biosynthesis and/or metabolism. For example, BPA can bind multiple oestrogen receptor subtypes. Moreover, several studies indicate that BPA also promotes signalling via extranuclear signal transduction pathways, including extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) and Akt [32], and BPA also exerts oestradiol-like effects by binding to ERα and thereby activating extranuclear pathways [33]. Thus, it is increasingly clear that the diverse effects of BPA are likely to arise from interactions across multiple signal transduction pathways [34]. Adding complexity is the fact that epidemiological data have linked BPA to disruptions in androgen-dependent developmental outcomes [35], despite few mechanistic data demonstrating that BPA alters androgen receptor activity or interferes with androgen-dependent extranuclear receptors. Finally, there is some evidence that BPA may also disrupt glucocorticoid receptor signalling [36,37,38]. Because endocrine signalling adapts to tonic activation through feedback circuits and because metabolic phenotypes arise from the integrated crosstalk of multiple signalling cascades that act additively, antagonistically or synergistically, the diversity of BPA actions illustrates how this and other EDCs may exhibit non-monotonic dose–response relationships in which phenotypic changes at low doses are not necessarily observed with higher exposures.
While significant literature linking developmental EDC exposures to metabolic disorders focuses on oestrogen receptor actions, robust data also indicate that perinatal exposures to some EDCs alter the expression or activity of the nuclear receptor peroxisome proliferator-activated receptor γ (PPARγ) and its heterodimeric partner retinoid X receptor (RXR), which play essential roles in adipocyte development and function. Interestingly, BPA has been shown to increase PPARγ expression [39]. While no studies have shown that BPA directly binds to PPARγ, brominated BPA analogues, also found in high quantities in the environment, bind to the receptor at low concentrations [40]. The prototypical obesogen TBT binds and activates both PPARγ and RXR at very low concentrations, augmenting adipocyte differentiation, promoting adipocyte dysfunction and resulting in transgenerational disruptions in metabolic homeostasis [26, 31, 41,42,43].
Our recent studies suggest that mitochondrial dysfunction could be an early event in the BPA-induced impairment of beta cells [18, 19]. Low-dose exposure of pregnant dams impairs beta cell mitochondria function, blunts GSIS, and reduces beta cell mass in male offspring [18]. These effects persisted into the second, but not the third, generation [19]. In another study, perinatal BPA exposure decreased activity of Complexes I and III of the electron transport chain in the livers of suckling rats, with later-life development of microvesicular liver steatosis and enhanced lipogenic gene expression [44]. Beyond BPA, other common EDCs have also been shown to disrupt mitochondrial function after developmental exposure, including arsenic [45].
Collectively, these data underscore the complexity of endocrine disruption with regard to metabolic outcomes. Unlike pharmaceutical agents that are designed to hit specific molecular targets, EDCs often interfere with multiple pathways, with specific effects likely dependent on both the dose and timing of exposure as well as the target and the background hormonal milieu. However, to date, while multiple studies have demonstrated that EDCs exhibit multiple modes of action, very few studies have definitively ascribed specific mechanisms to particular phenotypes. This greatly complicates our understanding of the molecular mechanisms of these agents and argues for comprehensive unbiased approaches for uncovering the network of metabolic toxicities induced by these agents.
Epigenetic mechanisms
With studies demonstrating transgenerational effects of EDCs on metabolic health, attention has turned to how EDCs directly influence the epigenome. Multiple studies report associations between developmental exposures to EDCs and epigenetic changes in key metabolic tissues, including beta cells, liver, adipose and muscle [7]. One of the earliest studies examining this phenomenon demonstrated that developmental BPA exposure shifts the coat colour of Agouti mice through stable epigenetic changes [46]. In general, imprinted genes are particularly vulnerable to de novo epigenetic modifications. Dams exposed to BPA from 2 weeks prior to mating to the end of gestation yielded offspring with loss of imprinting and biparental expression of Igf2, increased embryonic Igf2 gene expression and enhanced methylation of the Igf2 differentially methylated region 1 (DMR1) [17]. Extension of these studies demonstrated that Igf2 DMR1 hypermethylation persisted in islets of F1 and F2 offspring of BPA-exposed dams with an associated increased expression of Igf2 [18]. While Igf2 has multiple functions, it is a key regulator of beta cell development, and aberrant early-life Igf2 imprinting could potentially disrupt beta cell development.
Epigenetic alterations have also been described for several other metabolically active EDCs [7]. Phthalates are a family of phthalic acid diesters that are commonly found in consumer products, resulting in ubiquitous exposure in the USA. Limited but growing evidence in human studies indicate that phthalate exposure is associated with DNA methylation changes of imprinted genes in cord blood and placenta [47]. Animal studies demonstrate associations between phthalates and global and site-specific methylation [48]; however, studies were performed in mixed cell populations, and changes in DNA methylation were usually modest.
In addition to BPA and phthalates, other EDCs linked to epigenetic modifications include the metabolism-disrupting chemicals arsenic [49] and TBT [26]. Importantly, epigenetic modifications are cell- and tissue-specific, and findings from one tissue or cell type may not serve as a proxy for another. Moreover, even single cell types are often heterogeneous, and this lack of uniformity is likely to reflect differences in cell state or cell-specific functions. For example, not all beta cells function similarly in an islet, and this may be reflected in epigenetic differences. Demonstrating that epigenetic modifications induce changes in gene expression that, in turn, cause abnormal phenotypes is experimentally challenging; however, it is imperative to link exposure-associated changes to metabolic dysregulation.
Beyond offspring: impact of gestational exposures on mothers
While environmental exposures during gestation and lactation can clearly impact the long-term metabolic trajectory of offspring, the consequences of exposures during this window on mothers is less appreciated. Gestational diabetes (GDM) imposes unique pregnancy-associated risks, including gestational hypertension, preeclampsia, future diabetes and obstetric complications. Assessments of EDC effects on GDM in humans remain in their infancy; however, emerging data suggest potential associations. Commonly found in cosmetics, pharmaceuticals and food, parabens were associated with glucose levels during pregnancy in a cohort of women at high risk of GDM [50], while the widely used antibacterial triclosan was nearly significantly associated with GDM in another cohort [51]. In the Infant Development and Environment Study, monoethyl phthalate levels were associated with increased GDM risk [52]. Among the inorganic EDCs, arsenic has been associated with GDM in a host of studies. In the Maternal-Infant Research on Environmental Chemicals Study, the risk of GDM was nearly fourfold greater in the highest vs lowest tertile of dimethylarsinic acid levels [53]. Overall, arsenic has been associated with GDM in many [54,55,56,57] but not all studies [58]. In addition to disruptions during gestation, the dynamic changes that happen in the pancreatic islet under the influence of pregnancy hormones suggest that environmental insults that occur during this window may permanently reprogram beta cells and thereby alter their long-term function. Indeed, gestational BPA exposure was shown to not only induce metabolic dysfunction in adult offspring [59], but exposed dams also exhibited glucose intolerance later in life [60]. While more studies that employ comprehensive exposure assessments throughout pregnancy are needed, these data highlight pregnancy as a unique window of susceptibility to EDC-induced metabolic dysfunction.
Exposures in later life and metabolic dysfunction
Robust evidence now links exposure to a variety of EDCs with diabetes risk (reviewed extensively in refs. [7,8,9,10]). The majority of cross-sectional and prospective epidemiological analyses have been conducted in adult populations, while most animal studies have employed exposure paradigms using adult mice. Across these studies, there is great diversity in the spectrum of environmental toxicants linked to diabetes, with multiple potential mechanisms of metabolic toxicity (Fig. 2). EDCs linked to diabetes and/or alterations in insulin–glucose dynamics in human studies include arsenic, BPA, dioxins, organochlorine pesticides, PCBs and phthalates, among others [61,62,63,64,65,66,67,68,69,70,71,72]. In animal studies, disruptions in glucose homeostasis have been linked to arsenic, atrazine, BPA, cadmium, dioxins, malathion, particulate matter air pollution, PCBs, phthalates, persistent organic pollutants, tolylfluanid, and TBT [73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93]. Importantly, the diversity of EDCs now linked with metabolic dysfunction, coupled with the frequency with which human populations are exposed, now raise important questions with regard to how these diabetogenic agents interact with each other, as well as with traditional diabetes risk factors, to augment disease risk [94].
Indeed, diabetogenic EDC exposures do not exist within a vacuum; rather, exposures occur concurrently with other metabolic stressors. Recent animal models demonstrate that excess energy intake induced by provision of a high-fat or high-fat/high-sucrose diet augments the metabolic dysfunction induced by BPA [90, 95, 96], dichlorodiphenyltrichloroethane (DDT) [97], perfluoroalkyl substances [98], tolylfluanid [79] and phthalates [99]. Intriguingly, some studies suggest an EDC–diet potentiation that evolves over time as with dichlorodiphenyldichloroethylene (DDE) [100]. In contrast, other data suggest that changes in diet may precipitate adverse EDC effects; this has been suggested for PCBs, which may specifically impair glucose tolerance after diet-induced weight loss [91, 101]. Importantly, since many EDCs are lipophilic, high-fat diets are likely to augment exposure, whereas reductions in adipose mass with weight loss release EDCs into the circulation [102,103,104,105]. Whether this mobilisation from adipose tissue antagonises subsequent weight loss remains under debate.
Environmental diabetogens and health justice
It has long been recognised that diabetes disproportionately afflicts racial and ethnic minorities, as well as those with lower incomes. Despite this knowledge, these disparities have been amplified over time [106]. In addition to higher rates of disease, African-Americans and Latinx/Hispanics are more likely to suffer microvascular complications, including nephropathy [107], neuropathy/amputations [108] and retinopathy [109]. Furthermore, diabetes-related mortality is also higher in these communities [110]. While various social factors are posited to contribute to diabetes disparities, less appreciated is the additional contribution of environmental injustice to this heightened risk. In our recent analysis, five classes of EDCs associated with diabetic phenotypes in cell-based and animal studies (PCBs, organochlorine pesticides, chemical constituents of air pollution, BPA and phthalates) were shown to be linked with incident diabetes in prospective epidemiological studies [111]. Importantly, for each of these EDC classes, data indicated that racial and ethnic minorities, as well as those with lower incomes, are more highly exposed [111]. In addition, studies suggest that racial and ethnic minorities may be at heightened risk for EDC-induced metabolic dysfunction. In a meta-analysis, exposure to higher vs lower levels of both PCBs and organochlorine pesticides was associated with greater diabetes risk in non-white compared with white populations [65]. In a study of phthalates and metabolic control, phthalate-associated insulin resistance was observed in Mexican Americans and African-Americans but not in non-Hispanic whites [112]. In another study, phthalates were associated with worse glucose control and more severe insulin resistance in African-Americans and Mexican Americans compared with non-Hispanic whites [113]. A recent analysis examining the links between phthalates and GDM suggested potentially higher phthalate-associated risk among Asians [52]. Finally, in a study of overall mortality, the hazard ratio for death per unit increase in particulate matter air pollution was higher in Hispanics and much greater in blacks than in whites [114]. Whether the enhanced diabetes- and mortality-associated risks of environmental toxicants in communities with heightened diabetes risk arises from higher baseline exposures, gene–environment interactions or environment-amplified social risk factors remains to be clarified; however, regardless of the origin of this enhanced risk, addressing environmental potentiators of diabetes is essential for reducing disease disparities and improving health equity.
From abdication to engagement
With mounting evidence that EDCs contribute to diabetes pathogenesis, it is incumbent upon the diabetes care community to address the environmental drivers of the disease; however, current clinical practice guidelines wholly neglect environmental toxicants as diabetes risk factors [115]. While data are required to definitively demonstrate that exposure reduction improves outcomes, emerging evidence provides a framework for action [116]. Importantly, paradigms for environmental risk reduction are available in other fields, such as chronic obstructive lung disease [117] and asthma [118], which acknowledge the health threat posed by air pollution, an exposure also linked to diabetes [119, 120]. In addition, leadership from the American College of Obstetricians and Gynecologists, International Federation of Gynecology and Obstetrics, and the Endocrine Society provide further guidance on areas for risk reduction [5, 121, 122]. The urgency for such action is underscored by data from the European Union estimating that five EDCs alone add €18–29 billion annually to obesity- and diabetes-associated healthcare costs [123]. Conversely, data from the Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) study indicate that 25% reductions in only four target compounds (PCB 153, monoethylphthalate, DDE and perfluorononanoic acid) would reduce diabetes prevalence by 13% in Europe (over 150,000 cases), with associated cost savings of €4.51 billion annually [124]. Given the narrow focus of these analyses relative to the total burden of human chemical exposure, reducing contact with diabetogenic chemicals has potential to substantially lower the individual and societal toll of diabetes and its complications. Capitalising on this, however, requires both improved clinical guidance, as well as transformations in public policy to incorporate diabetes and its associated costs into the development of environmental policies [125].
Challenges and controversies
Despite consensus statements regarding EDC risks from the Organisation for Economic Co-operation and Development in the European Union and the Endocrine Society, some controversies persist. These include robust discussion regarding methodologies used in biomonitoring studies, including the validity of ELISA for measuring BPA concentrations in bodily tissues. Importantly, more recent studies have employed more sensitive and reliable techniques to measure very low levels of EDCs. It has also been suggested that low levels of unconjugated BPA in bodily tissues and fluids arise from contamination of collection materials and/or deconjugation of BPA metabolites during storage. To address this concern, multiple human and animal studies have detected BPA in a variety of biological specimens using alternative methods, and storage vials are now BPA-free. Furthermore, high-quality animal studies are now designed to limit background EDC exposure from diet, caging, bedding and water bottles. The route of exposure used in animal studies also remains hotly debated. While differences in exposure routes can affect circulating BPA levels, several studies have shown that there are fewer differences in route-dependent BPA metabolism and excretion than previously thought [126]. Collectively, these issues mandate rigorously designed epidemiological and animal studies. Importantly, recent clinical studies have demonstrated that administering BPA to humans alters glucose–insulin homeostasis [127, 128]. While these data validate the robust cell-based, animal and epidemiological evidence linking BPA to metabolic disruptions, they also raise an important ethical question: Will such direct evidence of harm be required for each of the thousands of chemicals to which humans are exposed before action is taken to protect public health?
Conclusions
Burgeoning evidence now implicates exposure to a variety of environmental toxicants in the pathogenesis of diabetes. These exposures occur across the lifespan; however, certain developmental periods are uniquely sensitive windows during which metabolism can be permanently disrupted in both the exposed individual and subsequent generations. Importantly, several communities at heightened diabetes risk are also exposed to higher levels of chemicals linked to the disease. Despite this evidence, clinical practice remains blind to EDCs as diabetes drivers. While more work is required to address knowledge gaps regarding environmental exposures and diabetes risk, the weight of the evidence now mandates action to empower individuals and governments to address environmental contributors to diabetes risk.
Abbreviations
- BPA:
-
Bisphenol A
- DMR1:
-
Differentially methylated region 1
- EDC:
-
Endocrine-disrupting chemical
- GDM:
-
Gestational diabetes
- GSIS:
-
Glucose-stimulated insulin secretion
- PCBs:
-
Polychlorinated biphenyls
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- RXR:
-
Retinoid X receptor
- TBT:
-
Tributyltin
References
International Diabetes Federation (2017) IDF Diabetes Atlas, 8th Edition. Available from www.idf.org/e-library/epidemiology-research/diabetes-atlas/134-idf-diabetes-atlas-8th-edition.html. Accessed 15 Jan 2018
World Health Organization (2016) Global report on diabetes. World Health Organization, Geneva
American Diabetes Association (2018) Economic costs of diabetes in the U.S. in 2017. Diabetes Care 41(5):917–928. https://doi.org/10.2337/dci18-0007
Squires E, Duber H, Campbell M et al (2018) Health care spending on diabetes in the U.S., 1996-2013. Diabetes Care 41(7):1423–1431. https://doi.org/10.2337/dc17-1376
Gore AC, Chappell VA, Fenton SE et al (2015) EDC-2: the Endocrine Society’s second scientific statement on endocrine-disrupting chemicals. Endocr Rev 36(6):E1–E150. https://doi.org/10.1210/er.2015-1010
Zoeller RT, Brown TR, Doan LL et al (2012) Endocrine-disrupting chemicals and public health protection: a statement of principles from the Endocrine Society. Endocrinology 153(9):4097–4110. https://doi.org/10.1210/en.2012-1422
Heindel JJ, Blumberg B, Cave M et al (2017) Metabolism disrupting chemicals and metabolic disorders. Reprod Toxicol 68:3–33. https://doi.org/10.1016/j.reprotox.2016.10.001
Mimoto MS, Nadal A, Sargis RM (2017) Polluted pathways: mechanisms of metabolic disruption by endocrine disrupting chemicals. Curr Environ Health Rep 4(2):208–222. https://doi.org/10.1007/s40572-017-0137-0
Sargis RM (2014) The hijacking of cellular signaling and the diabetes epidemic: mechanisms of environmental disruption of insulin action and glucose homeostasis. Diabetes Metab J 38(1):1–12. https://doi.org/10.4093/dmj.2014.38.1.1
Neel BA, Sargis RM (2011) The paradox of progress: environmental disruption of metabolism and the diabetes epidemic. Diabetes 60(7):1838–1848. https://doi.org/10.2337/db11-0153
Tuduri E, Marroqui L, Dos Santos RS, Quesada I, Fuentes E, Alonso-Magdalena P (2019) Timing of exposure to bisphenol-A: implications for diabetes development. Front Endocrinol (Lausanne) 9:648. https://doi.org/10.3389/fendo.2018.00648
Calafat AM, Ye X, Wong LY, Reidy JA, Needham LL (2008) Exposure of the U.S. population to bisphenol A and 4-tertiary-octylphenol: 2003-2004. Environ Health Perspect 116(1):39–44. https://doi.org/10.1289/ehp.10753
Ikezuki Y, Tsutsumi O, Takai Y, Kamei Y, Taketani Y (2002) Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Hum Reprod 17(11):2839–2841. https://doi.org/10.1093/humrep/17.11.2839
Schonfelder G, Wittfoht W, Hopp H, Talsness CE, Paul M, Chahoud I (2002) Parent bisphenol A accumulation in the human maternal-fetal-placental unit. Environ Health Perspect 110(11):A703–A707. https://doi.org/10.1289/ehp.110-1241091
Nahar MS, Liao C, Kannan K, Dolinoy DC (2013) Fetal liver bisphenol A concentrations and biotransformation gene expression reveal variable exposure and altered capacity for metabolism in humans. J Biochem Mol Toxicol 27(2):116–123. https://doi.org/10.1002/jbt.21459
Vom Saal FS, Nagel SC, Coe BL, Angle BM, Taylor JA (2012) The estrogenic endocrine disrupting chemical bisphenol A (BPA) and obesity. Mol Cell Endocrinol 354(1–2):74–84. https://doi.org/10.1016/j.mce.2012.01.001
Susiarjo M, Xin F, Bansal A et al (2015) Bisphenol A exposure disrupts metabolic health across multiple generations in the mouse. Endocrinology 156(6):2049–2058. https://doi.org/10.1210/en.2014-2027
Bansal A, Rashid C, Xin F et al (2017) Sex- and dose-specific effects of maternal bisphenol A exposure on pancreatic islets of first- and second-generation adult mice offspring. Environ Health Perspect 125(9):097022. https://doi.org/10.1289/EHP1674
Bansal A, Li C, Xin F, et al. (2018) Transgenerational effects of maternal bisphenol: a exposure on offspring metabolic health. J Dev Orig Health Dis 1–12. https://doi.org/10.1017/S2040174418000764
Rubin BS, Schaeberle CM, Soto AM (2019) The case for BPA as an obesogen: contributors to the controversy. Front Endocrinol (Lausanne) 10:30. https://doi.org/10.3389/fendo.2019.00030
Rubin BS, Murray MK, Damassa DA, King JC, Soto AM (2001) Perinatal exposure to low doses of bisphenol A affects body weight, patterns of estrous cyclicity, and plasma LH levels. Environ Health Perspect 109(7):675–680. https://doi.org/10.1289/ehp.01109675
Tiano JP, Mauvais-Jarvis F (2012) Importance of oestrogen receptors to preserve functional beta-cell mass in diabetes. Nat Rev Endocrinol 8(6):342–351. https://doi.org/10.1038/nrendo.2011.242
Whitehead R, Guan H, Arany E, Cernea M, Yang K (2016) Prenatal exposure to bisphenol A alters mouse fetal pancreatic morphology and islet composition. Horm Mol Biol Clin Invest 25(3):171–179. https://doi.org/10.1515/hmbci-2015-0052
Garcia-Arevalo M, Alonso-Magdalena P, Rebelo Dos Santos J, Quesada I, Carneiro EM, Nadal A (2014) Exposure to bisphenol-A during pregnancy partially mimics the effects of a high-fat diet altering glucose homeostasis and gene expression in adult male mice. PLoS One 9(6):e100214. https://doi.org/10.1371/journal.pone.0100214
Garcia-Arevalo M, Alonso-Magdalena P, Servitja JM et al (2016) Maternal exposure to bisphenol-A during pregnancy increases pancreatic beta-cell growth during early life in male mice offspring. Endocrinology 157(11):4158–4171. https://doi.org/10.1210/en.2016-1390
Chamorro-Garcia R, Diaz-Castillo C, Shoucri BM et al (2017) Ancestral perinatal obesogen exposure results in a transgenerational thrifty phenotype in mice. Nat Commun 8(1):2012. https://doi.org/10.1038/s41467-017-01944-z
Veiga-Lopez A, Pu Y, Gingrich J, Padmanabhan V (2018) Obesogenic endocrine disrupting chemicals: identifying knowledge gaps. Trends Endocrinol Metab 29(9):607–625. https://doi.org/10.1016/j.tem.2018.06.003
Mennigen JA, Thompson LM, Bell M, Tellez Santos M, Gore AC (2018) Transgenerational effects of polychlorinated biphenyls: 1. Development and physiology across 3 generations of rats. Environ Health 17(1):18. https://doi.org/10.1186/s12940-018-0362-5
Skinner MK (2016) Endocrine disruptors in 2015: epigenetic transgenerational inheritance. Nat Rev Endocrinol 12(2):68–70. https://doi.org/10.1038/nrendo.2015.206
Drobna Z, Henriksen AD, Wolstenholme JT et al (2018) Transgenerational effects of bisphenol A on gene expression and DNA methylation of imprinted genes in brain. Endocrinology 159(1):132–144. https://doi.org/10.1210/en.2017-00730
Chamorro-Garcia R, Sahu M, Abbey RJ, Laude J, Pham N, Blumberg B (2013) Transgenerational inheritance of increased fat depot size, stem cell reprogramming, and hepatic steatosis elicited by prenatal exposure to the obesogen tributyltin in mice. Environ Health Perspect 121(3):359–366. https://doi.org/10.1289/ehp.1205701
Ptak A, Gregoraszczuk EL (2012) Bisphenol A induces leptin receptor expression, creating more binding sites for leptin, and activates the JAK/Stat, MAPK/ERK and PI3K/Akt signalling pathways in human ovarian cancer cell. Toxicol Lett 210(3):332–337. https://doi.org/10.1016/j.toxlet.2012.02.003
Xu X, Lu Y, Zhang G et al (2014) Bisphenol A promotes dendritic morphogenesis of hippocampal neurons through estrogen receptor-mediated ERK1/2 signal pathway. Chemosphere 96:129–137. https://doi.org/10.1016/j.chemosphere.2013.09.063
Acconcia F, Pallottini V, Marino M (2015) Molecular mechanisms of action of BPA. Dose Response 13(4):1559325815610582. https://doi.org/10.1177/1559325815610582
Fernandez MF, Arrebola JP, Jimenez-Diaz I et al (2016) Bisphenol A and other phenols in human placenta from children with cryptorchidism or hypospadias. Reprod Toxicol 59:89–95. https://doi.org/10.1016/j.reprotox.2015.11.002
Prasanth GK, Divya LM, Sadasivan C (2010) Bisphenol-A can bind to human glucocorticoid receptor as an agonist: an in silico study. J Appl Toxicol 30(8):769–774. https://doi.org/10.1002/jat.1570
Sargis RM, Johnson DN, Choudhury RA, Brady MJ (2010) Environmental endocrine disruptors promote adipogenesis in the 3T3-L1 cell line through glucocorticoid receptor activation. Obesity 18(7):1283–1288. https://doi.org/10.1038/oby.2009.419
Wang J, Sun B, Hou M, Pan X, Li X (2013) The environmental obesogen bisphenol A promotes adipogenesis by increasing the amount of 11β-hydroxysteroid dehydrogenase type 1 in the adipose tissue of children. Int J Obes 37(7):999–1005. https://doi.org/10.1038/ijo.2012.173
Somm E, Schwitzgebel VM, Toulotte A et al (2009) Perinatal exposure to bisphenol A alters early adipogenesis in the rat. Environ Health Perspect 117(10):1549–1555. https://doi.org/10.1289/ehp.11342
Riu A, Grimaldi M, le Maire A et al (2011) Peroxisome proliferator-activated receptor gamma is a target for halogenated analogs of bisphenol A. Environ Health Perspect 119(9):1227–1232. https://doi.org/10.1289/ehp.1003328
Grun F, Watanabe H, Zamanian Z et al (2006) Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol Endocrinol 20(9):2141–2155. https://doi.org/10.1210/me.2005-0367
Shoucri BM, Hung VT, Chamorro-Garcia R, Shioda T, Blumberg B (2018) Retinoid X receptor activation during adipogenesis of female mesenchymal stem cells programs a dysfunctional adipocyte. Endocrinology 159(8):2863–2883. https://doi.org/10.1210/en.2018-00056
Regnier SM, El-Hashani E, Kamau W, Zhang X, Massad NL, Sargis RM (2015) Tributyltin differentially promotes development of a phenotypically distinct adipocyte. Obesity 23(9):1864–1871. https://doi.org/10.1002/oby.21174
Jiang Y, Xia W, Zhu Y et al (2014) Mitochondrial dysfunction in early life resulted from perinatal bisphenol A exposure contributes to hepatic steatosis in rat offspring. Toxicol Lett 228(2):85–92. https://doi.org/10.1016/j.toxlet.2014.04.013
Chandravanshi LP, Gupta R, Shukla RK (2018) Developmental neurotoxicity of arsenic: involvement of oxidative stress and mitochondrial functions. Biol Trace Elem Res 186(1):185–198. https://doi.org/10.1007/s12011-018-1286-1
Dolinoy DC, Huang D, Jirtle RL (2007) Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A 104(32):13056–13061. https://doi.org/10.1073/pnas.0703739104
Tindula G, Murphy SK, Grenier C et al (2018) DNA methylation of imprinted genes in Mexican-American newborn children with prenatal phthalate exposure. Epigenomics 10(7):1011–1026. https://doi.org/10.2217/epi-2017-0178
Pogribny IP, Tryndyak VP, Boureiko A et al (2008) Mechanisms of peroxisome proliferator-induced DNA hypomethylation in rat liver. Mutat Res 644(1–2):17–23. https://doi.org/10.1016/j.mrfmmm.2008.06.009
Meakin CJ, Martin EM, Szilagyi JT, Nylander-French LA, Fry RC (2019) Inorganic arsenic as an endocrine disruptor: modulation of the glucocorticoid receptor pathway in placental cells via CpG methylation. Chem Res Toxicol 32(3):493–499. https://doi.org/10.1021/acs.chemrestox.8b00352
Bellavia A, Chiu YH, Brown FM et al (2019) Urinary concentrations of parabens mixture and pregnancy glucose levels among women from a fertility clinic. Environ Res 168:389–396. https://doi.org/10.1016/j.envres.2018.10.009
Ouyang F, Tang N, Zhang HJ et al (2018) Maternal urinary triclosan level, gestational diabetes mellitus and birth weight in Chinese women. Sci Total Environ 626:451–457. https://doi.org/10.1016/j.scitotenv.2018.01.102
Shaffer RM, Ferguson KK, Sheppard L et al (2019) Maternal urinary phthalate metabolites in relation to gestational diabetes and glucose intolerance during pregnancy. Environ Int 123:588–596. https://doi.org/10.1016/j.envint.2018.12.021
Ashley-Martin J, Dodds L, Arbuckle TE et al (2018) Association between maternal urinary speciated arsenic concentrations and gestational diabetes in a cohort of Canadian women. Environ Int 121(Pt 1):714–720. https://doi.org/10.1016/j.envint.2018.10.008
Xia X, Liang C, Sheng J et al (2018) Association between serum arsenic levels and gestational diabetes mellitus: a population-based birth cohort study. Environ Pollut 235:850–856. https://doi.org/10.1016/j.envpol.2018.01.016
Marie C, Leger S, Guttmann A et al (2018) Exposure to arsenic in tap water and gestational diabetes: a French semi-ecological study. Environ Res 161:248–255. https://doi.org/10.1016/j.envres.2017.11.016
Farzan SF, Gossai A, Chen Y, Chasan-Taber L, Baker E, Karagas M (2016) Maternal arsenic exposure and gestational diabetes and glucose intolerance in the New Hampshire birth cohort study. Environ Health 15(1):106. https://doi.org/10.1186/s12940-016-0194-0
Shapiro GD, Dodds L, Arbuckle TE et al (2015) Exposure to phthalates, bisphenol A and metals in pregnancy and the association with impaired glucose tolerance and gestational diabetes mellitus: the MIREC study. Environ Int 83:63–71. https://doi.org/10.1016/j.envint.2015.05.016
Munoz MP, Valdes M, Munoz-Quezada MT et al (2018) Urinary inorganic arsenic concentration and gestational diabetes mellitus in pregnant women from Arica, Chile. Int J Environ Res Public Health 15(7). https://doi.org/10.3390/ijerph15071418
Alonso-Magdalena P, Vieira E, Soriano S et al (2010) Bisphenol A exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring. Environ Health Perspect 118(9):1243–1250. https://doi.org/10.1289/ehp.1001993
Alonso-Magdalena P, Garcia-Arevalo M, Quesada I, Nadal A (2015) Bisphenol-A treatment during pregnancy in mice: a new window of susceptibility for the development of diabetes in mothers later in life. Endocrinology 156(5):1659–1670. https://doi.org/10.1210/en.2014-1952
Maull EA, Ahsan H, Edwards J et al (2012) Evaluation of the association between arsenic and diabetes: a National Toxicology Program workshop review. Environ Health Perspect 120(12):1658–1670. https://doi.org/10.1289/ehp.1104579
Sung TC, Huang JW, Guo HR (2015) Association between arsenic exposure and diabetes: a meta-analysis. Biomed Res Int 2015:368087–368010. https://doi.org/10.1155/2015/368087
Wang W, Xie Z, Lin Y, Zhang D (2014) Association of inorganic arsenic exposure with type 2 diabetes mellitus: a meta-analysis. J Epidemiol Community Health 68(2):176–184. https://doi.org/10.1136/jech-2013-203114
Ranciere F, Lyons JG, Loh VH et al (2015) Bisphenol A and the risk of cardiometabolic disorders: a systematic review with meta-analysis of the epidemiological evidence. Environ Health 14(1):46. https://doi.org/10.1186/s12940-015-0036-5
Song Y, Chou EL, Baecker A et al (2016) Endocrine-disrupting chemicals, risk of type 2 diabetes, and diabetes-related metabolic traits: a systematic review and meta-analysis. J Diabetes 8(4):516–532. https://doi.org/10.1111/1753-0407.12325
Jensen TK, Timmermann AG, Rossing LI et al (2014) Polychlorinated biphenyl exposure and glucose metabolism in 9-year-old Danish children. J Clin Endocrinol Metab 99(12):E2643–E2651. https://doi.org/10.1210/jc.2014-1683
Suarez-Lopez JR, Lee DH, Porta M, Steffes MW, Jacobs DR Jr (2015) Persistent organic pollutants in young adults and changes in glucose related metabolism over a 23-year follow-up. Environ Res 137:485–494. https://doi.org/10.1016/j.envres.2014.11.001
Tang M, Chen K, Yang F, Liu W (2014) Exposure to organochlorine pollutants and type 2 diabetes: a systematic review and meta-analysis. PLoS One 9(10):e85556. https://doi.org/10.1371/journal.pone.0085556
Taylor KW, Novak RF, Anderson HA et al (2013) Evaluation of the association between persistent organic pollutants (POPs) and diabetes in epidemiological studies: a national toxicology program workshop review. Environ Health Perspect 121(7):774–783. https://doi.org/10.1289/ehp.1205502
Shoshtari-Yeganeh B, Zarean M, Mansourian M et al (2019) Systematic review and meta-analysis on the association between phthalates exposure and insulin resistance. Environ Sci Pollut Res Int 26(10):9435–9442. https://doi.org/10.1007/s11356-019-04373-1
Sun Q, Cornelis MC, Townsend MK et al (2014) Association of urinary concentrations of bisphenol a and phthalate metabolites with risk of type 2 diabetes: a prospective investigation in the Nurses’ Health Study (NHS) and NHSII cohorts. Environ Health Perspect 122(6):616–623. https://doi.org/10.1289/ehp.1307201
Lind PM, Zethelius B, Lind L (2012) Circulating levels of phthalate metabolites are associated with prevalent diabetes in the elderly. Diabetes Care 35(7):1519–1524. https://doi.org/10.2337/dc11-2396
Rajesh P, Sathish S, Srinivasan C, Selvaraj J, Balasubramanian K (2013) Phthalate is associated with insulin resistance in adipose tissue of male rat: role of antioxidant vitamins. J Cell Biochem 114(3):558–569. https://doi.org/10.1002/jcb.24399
Indumathi D, Jayashree S, Selvaraj J et al (2013) Effect of bisphenol-A on insulin signal transduction and glucose oxidation in skeletal muscle of adult male albino rat. Hum Exp Toxicol 32(9):960–971. https://doi.org/10.1177/0960327112470273
Jayashree S, Indumathi D, Akilavalli N, Sathish S, Selvaraj J, Balasubramanian K (2013) Effect of bisphenol-A on insulin signal transduction and glucose oxidation in liver of adult male albino rat. Environ Toxicol Pharmacol 35(2):300–310. https://doi.org/10.1016/j.etap.2012.12.016
Han JC, Park SY, Hah BG et al (2003) Cadmium induces impaired glucose tolerance in rat by down-regulating GLUT4 expression in adipocytes. Arch Biochem Biophys 413(2):213–220. https://doi.org/10.1016/S0003-9861(03)00120-6
Liu PC, Matsumura F (1995) Differential effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the "adipose- type" and "brain-type" glucose transporters in mice. Mol Pharmacol 47(1):65–73
Enan E, Matsumura F (1994) 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)-induced changes in glucose transporting activity in guinea pigs, mice, and rats in vivo and in vitro. J Biochem Toxicol 9(2):97–106. https://doi.org/10.1002/jbt.2570090207
Regnier SM, Kirkley AG, Ruiz D et al (2018) Diet-dependence of metabolic perturbations mediated by the endocrine disruptor tolylfluanid. Endocr Connect 7(1):159–168. https://doi.org/10.1530/EC-17-0320
Regnier SM, Kirkley AG, Ye H et al (2015) Dietary exposure to the endocrine disruptor tolylfluanid promotes global metabolic dysfunction in male mice. Endocrinology 156(3):896–910. https://doi.org/10.1210/en.2014-1668
Paul DS, Walton FS, Saunders RJ, Styblo M (2011) Characterization of the impaired glucose homeostasis produced in C57BL/6 mice by chronic exposure to arsenic and high-fat diet. Environ Health Perspect 119(8):1104–1109. https://doi.org/10.1289/ehp.1003324
Kirkley AG, Carmean CM, Ruiz D et al (2018) Arsenic exposure induces glucose intolerance and alters global energy metabolism. Am J Physiol Regul Integr Comp Physiol 314(2):R294–R303. https://doi.org/10.1152/ajpregu.00522.2016
Hill DS, Wlodarczyk BJ, Mitchell LE, Finnell RH (2009) Arsenate-induced maternal glucose intolerance and neural tube defects in a mouse model. Toxicol Appl Pharmacol 239(1):29–36. https://doi.org/10.1016/j.taap.2009.05.009
Sun Q, Yue P, Deiuliis JA et al (2009) Ambient air pollution exaggerates adipose inflammation and insulin resistance in a mouse model of diet-induced obesity. Circulation 119(4):538–546. https://doi.org/10.1161/CIRCULATIONAHA.108.799015
Panahi P, Vosough-Ghanbari S, Pournourmohammadi S et al (2006) Stimulatory effects of malathion on the key enzymes activities of insulin secretion in langerhans islets, glutamate dehydrogenase and glucokinase. Toxicol Mech Methods 16(4):161–167. https://doi.org/10.1080/15376520500191623
Batista TM, Alonso-Magdalena P, Vieira E et al (2012) Short-term treatment with bisphenol-a leads to metabolic abnormalities in adult male mice. PLoS One 7(3):e33814. https://doi.org/10.1371/journal.pone.0033814
Zuo Z, Chen S, Wu T et al (2011) Tributyltin causes obesity and hepatic steatosis in male mice. Environ Toxicol 26(1):79–85. https://doi.org/10.1002/tox.20531
Huang CF, Yang CY, Chan DC et al (2015) Arsenic exposure and glucose intolerance/insulin resistance in estrogen-deficient female mice. Environ Health Perspect 123(11):1138–1144. https://doi.org/10.1289/ehp.1408663
Lim S, Ahn SY, Song IC et al (2009) Chronic exposure to the herbicide, atrazine, causes mitochondrial dysfunction and insulin resistance. PLoS One 4(4):e5186. https://doi.org/10.1371/journal.pone.0005186
Moon MK, Jeong IK, Jung Oh T et al (2015) Long-term oral exposure to bisphenol A induces glucose intolerance and insulin resistance. J Endocrinol 226(1):35–42. https://doi.org/10.1530/JOE-14-0714
Baker NA, Karounos M, English V et al (2013) Coplanar polychlorinated biphenyls impair glucose homeostasis in lean C57BL/6 mice and mitigate beneficial effects of weight loss on glucose homeostasis in obese mice. Environ Health Perspect 121(1):105–110. https://doi.org/10.1289/ehp.1205421
Gray SL, Shaw AC, Gagne AX, Chan HM (2013) Chronic exposure to PCBs (Aroclor 1254) exacerbates obesity-induced insulin resistance and hyperinsulinemia in mice. J Toxicol Environ Health A 76(12):701–715. https://doi.org/10.1080/15287394.2013.796503
Huang CF, Yang CY, Tsai JR, Wu CT, Liu SH, Lan KC (2018) Low-dose tributyltin exposure induces an oxidative stress-triggered JNK-related pancreatic beta-cell apoptosis and a reversible hypoinsulinemic hyperglycemia in mice. Sci Rep 8(1):5734. https://doi.org/10.1038/s41598-018-24076-w
Sargis RM (2015) Metabolic disruption in context: clinical avenues for synergistic perturbations in energy homeostasis by endocrine disrupting chemicals. Endocrine Disruptors 3(1):e1080788-1080781-1080787. https://doi.org/10.1080/23273747.2015.1080788
Wei J, Lin Y, Li Y et al (2011) Perinatal exposure to bisphenol A at reference dose predisposes offspring to metabolic syndrome in adult rats on a high-fat diet. Endocrinology 152(8):3049–3061. https://doi.org/10.1210/en.2011-0045
Ding S, Fan Y, Zhao N et al (2014) High-fat diet aggravates glucose homeostasis disorder caused by chronic exposure to bisphenol A. J Endocrinol 221(1):167–179. https://doi.org/10.1530/JOE-13-0386
La Merrill M, Karey E, Moshier E et al (2014) Perinatal exposure of mice to the pesticide DDT impairs energy expenditure and metabolism in adult female offspring. PLoS One 9(7):e103337. https://doi.org/10.1371/journal.pone.0103337
Wan HT, Zhao YG, Leung PY, Wong CK (2014) Perinatal exposure to perfluorooctane sulfonate affects glucose metabolism in adult offspring. PLoS One 9(1):e87137. https://doi.org/10.1371/journal.pone.0087137
Strakovsky RS, Lezmi S, Shkoda I, Flaws JA, Helferich WG, Pan YX (2015) In utero growth restriction and catch-up adipogenesis after developmental di (2-ethylhexyl) phthalate exposure cause glucose intolerance in adult male rats following a high-fat dietary challenge. J Nutr Biochem 26(11):1208–1220. https://doi.org/10.1016/j.jnutbio.2015.05.012
Howell GE 3rd, Mulligan C, Meek E, Chambers JE (2015) Effect of chronic p,p'-dichlorodiphenyldichloroethylene (DDE) exposure on high fat diet-induced alterations in glucose and lipid metabolism in male C57BL/6H mice. Toxicology 328:112–122. https://doi.org/10.1016/j.tox.2014.12.017
Baker NA, Shoemaker R, English V et al (2015) Effects of adipocyte aryl hydrocarbon receptor deficiency on PCB-induced disruption of glucose homeostasis in lean and obese mice. Environ Health Perspect 123(10):944–950. https://doi.org/10.1289/ehp.1408594
Dirtu AC, Dirinck E, Malarvannan G et al (2013) Dynamics of organohalogenated contaminants in human serum from obese individuals during one year of weight loss treatment. Environ Sci Technol 47(21):12441–12449. https://doi.org/10.1021/es400657t
Dirinck E, Dirtu AC, Jorens PG, Malarvannan G, Covaci A, Van Gaal LF (2015) Pivotal role for the visceral fat compartment in the release of persistent organic pollutants during weight loss. J Clin Endocrinol Metab 100(12):4463–4471. https://doi.org/10.1210/jc.2015-2571
Hue O, Marcotte J, Berrigan F et al (2006) Increased plasma levels of toxic pollutants accompanying weight loss induced by hypocaloric diet or by bariatric surgery. Obes Surg 16(9):1145–1154. https://doi.org/10.1381/096089206778392356
Charlier C, Desaive C, Plomteux G (2002) Human exposure to endocrine disrupters: consequences of gastroplasty on plasma concentration of toxic pollutants. Int J Obes Relat Metab Disord 26(11):1465–1468. https://doi.org/10.1038/sj.ijo.0802144
Beckles GL, Chou CF, Centers for Disease Control and Prevention (CDC) (2013) Diabetes - United States, 2006 and 2010. MMWR Surveill Summ 62(3):99–104
Ferdinand KC, Nasser SA (2015) Racial/ethnic disparities in prevalence and care of patients with type 2 diabetes mellitus. Curr Med Res Opin 31(5):913–923. https://doi.org/10.1185/03007995.2015.1029894
Peek ME (2011) Gender differences in diabetes-related lower extremity amputations. Clin Orthop Relat Res 469(7):1951–1955. https://doi.org/10.1007/s11999-010-1735-4
Nsiah-Kumi P, Ortmeier SR, Brown AE (2009) Disparities in diabetic retinopathy screening and disease for racial and ethnic minority populations—a literature review. J Natl Med Assoc 101(5):430–437. https://doi.org/10.1016/S0027-9684(15)30929-9
Rosenstock S, Whitman S, West JF, Balkin M (2014) Racial disparities in diabetes mortality in the 50 most populous US cities. J Urban Health 91(5):873–885. https://doi.org/10.1007/s11524-013-9861-4
Ruiz D, Becerra M, Jagai JS, Ard K, Sargis RM (2018) Disparities in environmental exposures to endocrine-disrupting chemicals and diabetes risk in vulnerable populations. Diabetes Care 41(1):193–205. https://doi.org/10.2337/dc16-2765
Trasande L, Spanier AJ, Sathyanarayana S, Attina TM, Blustein J (2013) Urinary phthalates and increased insulin resistance in adolescents. Pediatrics 132(3):e646–e655. https://doi.org/10.1542/peds.2012-4022
Huang T, Saxena AR, Isganaitis E, James-Todd T (2014) Gender and racial/ethnic differences in the associations of urinary phthalate metabolites with markers of diabetes risk: National Health and Nutrition Examination Survey 2001-2008. Environ Health 13(1):6. https://doi.org/10.1186/1476-069X-13-6
Di Q, Wang Y, Zanobetti A et al (2017) Air pollution and mortality in the medicare population. N Engl J Med 376(26):2513–2522. https://doi.org/10.1056/NEJMoa1702747
American Diabetes Association (2019) Standards of medical care in diabetes: 2019. Diabetes Care 42(Suppl 1):S1–S186. https://doi.org/10.2337/dc19-S001
Sargis RM, Heindel JJ, Padmanabhan V (2019) Interventions to address environmental metabolism-disrupting chemicals: changing the narrative to empower action to restore metabolic health. Front Endocrinol (Lausanne) 10:33. https://doi.org/10.3389/fendo.2019.00033
Vogelmeier CF, Criner GJ, Martinez FJ et al (2017) Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. Am J Respir Crit Care Med 195(5):557–582. https://doi.org/10.1164/rccm.201701-0218PP
National Asthma Education Prevention Program (2007) Expert Panel Report 3 (EPR-3): Guidelines for the Diagnosis and Management of Asthma-Summary Report 2007. J Allergy Clin Immunol 120(5 Suppl):94–138. https://doi.org/10.1016/j.jaci.2007.09.043
Wang B, Xu D, Jing Z, Liu D, Yan S, Wang Y (2014) Effect of long-term exposure to air pollution on type 2 diabetes mellitus risk: a systemic review and meta-analysis of cohort studies. Eur J Endocrinol 171(5):R173–R182. https://doi.org/10.1530/EJE-14-0365
Zhao Z, Lin F, Wang B, Cao Y, Hou X, Wang Y (2016) Residential proximity to major roadways and risk of type 2 diabetes mellitus: a meta-analysis. Int J Environ Res Public Health 14(1). https://doi.org/10.3390/ijerph14010003
Di Renzo GC, Conry JA, Blake J et al (2015) International Federation of Gynecology and Obstetrics opinion on reproductive health impacts of exposure to toxic environmental chemicals. Int J Gynaecol Obstet 131(3):219–225. https://doi.org/10.1016/j.ijgo.2015.09.002
American College of Obstetricians and Gynecologists (2013) Exposure to toxic environmental agents. Fertil Steril 100(4):931–934. https://doi.org/10.1016/j.fertnstert.2013.08.043
Legler J, Fletcher T, Govarts E et al (2015) Obesity, diabetes, and associated costs of exposure to endocrine-disrupting chemicals in the European Union. J Clin Endocrinol Metab 100(4):1278–1288. https://doi.org/10.1210/jc.2014-4326
Trasande L, Lampa E, Lind L, Lind PM (2017) Population attributable risks and costs of diabetogenic chemical exposures in the elderly. J Epidemiol Community Health 71(2):111–114. https://doi.org/10.1136/jech-2016-208006
Shaikh S, Jagai JS, Ashley C, Zhou S, Sargis RM (2018) Underutilized and under threat: environmental policy as a tool to address diabetes risk. Curr Diab Rep 18(5):25. https://doi.org/10.1007/s11892-018-0993-5
Vandenberg LN, Maffini MV, Sonnenschein C, Rubin BS, Soto AM (2009) Bisphenol-A and the great divide: a review of controversies in the field of endocrine disruption. Endocr Rev 30(1):75–95. https://doi.org/10.1210/er.2008-0021
Stahlhut RW, Myers JP, Taylor JA, Nadal A, Dyer JA, Vom Saal FS (2018) Experimental BPA exposure and glucose-stimulated insulin response in adult men and women. J Endocr Soc 2(10):1173–1187. https://doi.org/10.1210/js.2018-00151
Hagobian TA, Bird A, Stanelle S, Williams D, Schaffner A, Phelan S (2019) Pilot study on the effect of orally administered bisphenol A on glucose and insulin response in nonobese adults. J Endocr Soc 3(3):643–654. https://doi.org/10.1210/js.2018-00322
Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444(7121):840–846. https://doi.org/10.1038/nature05482
Funding
Work in the authors’ laboratories is supported by the National Institutes of Health (R01 ES028879, P30 ES027792 and R01 ES025159 supporting RMS; R01 ES023284, R01 ES028206 and P30 ES013508 supporting RAS).
Author information
Authors and Affiliations
Contributions
Both authors were responsible for drafting the article and revising it critically for important intellectual content. Both authors approved the version to be published.
Corresponding author
Ethics declarations
RMS declares he has received honoraria from CVS/Health. RAS has no duality of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Slideset of figures
(PPTX 492 kb)
Rights and permissions
About this article

Cite this article
Sargis, R.M., Simmons, R.A. Environmental neglect: endocrine disruptors as underappreciated but potentially modifiable diabetes risk factors. Diabetologia 62, 1811–1822 (2019). https://doi.org/10.1007/s00125-019-4940-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-019-4940-z