
Abstract
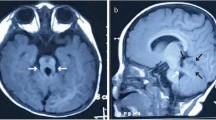
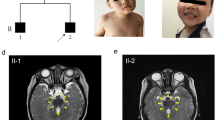
Joubert syndrome (JBTS), a rare genetic disorder resulted from primary cilium defects or basal-body dysfunction, is characterized by agenesis of cerebellar vermis and abnormal brain stem. Both genotypes and phenotypes of JBTS are highly heterogeneous. The identification of pathogenic gene variation is essential for making a definite diagnosis on JBTS. Here, we found that hypoplasia of cerebellar vermis occurred in three male members in a Chinese family. Then, we performed whole exome sequencing to identify a novel missense mutation c.599T > C (p. L200P) in the OFD1 gene which is the candidate gene of X-linked JBTS (JBST10). The following analysis showed that the variant was absent in the 1000 Genomes, ExAC and the 200 female controls; the position 200 Leucine residue was highly conserved across species; the missense variant was predicted to be deleterious using PolyPhen-2, PROVEAN, SIFT and Mutation Taster. The OFD1 expression was heavily lower in the proband and an induced male fetus compared with a healthy male with a wild-type OFD1 gene. The in vitro expression analysis of transiently transfecting c.599T or c.599C plasmids into HEK-293T cells confirmed that the missense mutation caused OFD1 reduction at the protein level. And further the mutated OFD1 decreased the level of Gli1 protein, a read-out of Sonic hedgehog (SHH) signaling essential for development of central neural system. A known pathogenic variant c.515T > C (p. L172P) showed the similar results. All of these observations suggested that the missense mutation causes the loss function of OFD1, resulting in SHH signaling impairs and brain development abnormality. In addition, the three patients have Dandy-Walker malformation, macrogyria and tetralogy of Fallot, respectively, the latter two of which are firstly found in JBTS10 patients. In conclusion, our findings expand the context of genotype and phenotype in the JBTS10 patients.




Similar content being viewed by others

Data availability
The raw WES data is deposited in BioProject database and the accession codes: PRJNA660941.
Code availability
Not applicable.
References
Bachmann-Gagescu R, Dempsey JC, Phelps IG, O'Roak BJ, Knutzen DM, Rue TC, Ishak GE, Isabella CR, Gorden N, Adkins J, Boyle EA, de Lacy N, O'Day D, Alswaid A, Ramadevi AR, Lingappa L, Lourenco C, Martorell L, Garcia-Cazorla A, Ozyurek H, Haliloglu G, Tuysuz B, Topcu M, Chance P, Parisi MA, Glass IA, Shendure J, Doherty D, University of Washington Center for Mendelian G (2015) Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet 52:514–522
Balaji NK, Sukumar IP, Cherian G (1977) Tetralogy of Fallot with polycystic kidneys. A case report. Indian Heart J 29:101–104
Bhardwaj P, Sharma M, Ahluwalia K (2018) Joubert syndrome with orofacial digital features. J Neurosci Rural Pract 9:152–154
Bouman A, Alders M, Oostra RJ, van Leeuwen E, Thuijs N, van der Kevie-Kersemaekers AM, van Maarle M (2017) Oral-facial-digital syndrome type 1 in males: congenital heart defects are included in its phenotypic spectrum. Am J Med Genet A 173:1383–1389
Coene KLM, Roepman R, Doherty D, Afroze B, Kroes HY, Letteboer SJF, Ngu LH, Budny B, van Wijk E, Gorden NT, Azhimi M, Thauvin-Robinet C, Veltman JA, Boink M, Kleefstra T, Cremers FPM, van Bokhoven H, de Brouwer APM (2009) OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am J Hum Genet 85:465–481
Field M, Scheffer IE, Gill D, Wilson M, Christie L, Shaw M, Gardner A, Glubb G, Hobson L, Corbett M, Friend K, Willis-Owen S, Gecz J (2012) Expanding the molecular basis and phenotypic spectrum of X-linked Joubert syndrome associated with OFD1 mutations. Eur J Hum Genet EJHG 20:806–809
Fleming LR, Doherty DA, Parisi MA, Glass IA, Bryant J, Fischer R, Turkbey B, Choyke P, Daryanani K, Vemulapalli M, Mullikin JC, Malicdan MC, Vilboux T, Sayer JA, Gahl WA, Gunay-Aygun M (2017) Prospective evaluation of kidney disease in Joubert syndrome. Clin J Am Soc Nephrol 12:1962–1973
Gerlitz G, Darhin E, Giorgio G, Franco B, Reiner O (2005) Novel functional features of the Lis-H domain: role in protein dimerization, half-life and cellular localization. Cell Cycle 4:1632–1640
Juric-Sekhar G, Adkins J, Doherty D, Hevner RF (2012) Joubert syndrome: brain and spinal cord malformations in genotyped cases and implications for neurodevelopmental functions of primary cilia. Acta Neuropathol 123:695–709
Lee J, Platt KA, Censullo P, Altaba ARI (1997) Gli1 is a target of Sonic hedgehog that induces ventral neural tube development. Development 124:2537–2552
Linpeng SY, Liu J, Pan JY, Cao YX, Teng YL, Liang DS, Li Z, Wu LQ (2018) Diagnosis of joubert syndrome 10 in a fetus with suspected Dandy-Walker variant by WES: a novel splicing mutation in OFD1. Biomed Res Int
Poretti A, Boltshauser E, Valente EM (2014) The molar tooth sign is pathognomonic for Joubert syndrome! Pediatr Neurol 50:e15–16
Romani M, Micalizzi A, Valente EM (2013) Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol 12:894–905
Sasai N, Briscoe J (2012) Primary cilia and graded Sonic Hedgehog signaling. Wiley Interdiscipl Rev Dev Biol 1:753–772
Singla V, Romaguera-Ros M, Garcia-Verdugo JM, Reiter JF (2010) Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev Cell 18:410–424
Slavotinek AM, Dutra A, Kpodzo D, Pak E, Nakane T, Turner J, Whiteford M, Biesecker LG, Stratton P (2004) A female with complete lack of Mullerian fusion, postaxial polydactyly, and tetralogy of Fallot: genetic heterogeneity of McKusick-Kaufman syndrome or a unique syndrome? Am J Med Genet A 129A:69–72
Strongin A, Heller T, Doherty D, Glass IA, Parisi MA, Bryant J, Choyke P, Turkbey B, Daryanani K, Yildirimli D, Vemulapalli M, Mullikin JC, Malicdan MC, Vilboux T, Gahl WA, Gunay-Aygun M, Progra NCS (2018) Characteristics of liver disease in 100 individuals with Joubert syndrome prospectively evaluated at a single center. J Pediatr Gastroenterol Nutr 66:428–435
Youn YH, Han YG (2018) Primary cilia in brain development and diseases. Am J Pathol 188:11–22
Acknowledgements
The authors would like to thank the professor Yang Xiaoyan of West China Second University Hospital for her contribution to this article.
Funding
This study was funded by National Natural Science Foundation of China with Grant (No. 81871203).
Author information
Authors and Affiliations
Contributions
YWZ, YQL and YY designed the study. YWZ investigated the subject and drafted this manuscript. HBQ analyzed image data. NL provided control samples. XYL was responsible for statistical analysis. YY revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This study was approved by the ethic committee of West China hospital and the Second Clinical Institute of North Sichuan Medical College.
Human and animal rights
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from the parents.
Consent for publication
Patients signed informed consent regarding publishing their data and photographs.
Additional information
Communicated by Stefan Hohmann.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, Yw., Qu, Hb., Long, N. et al. A rare mutant of OFD1 gene responsible for Joubert syndrome with significant phenotype variation. Mol Genet Genomics 296, 33–40 (2021). https://doi.org/10.1007/s00438-020-01726-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-020-01726-1