
Abstract
Ethylendiaminetetraacetic acid (EDTA) substituted and diethylenetriaminopentaacetic acid (DTPA) substituted aminated free-base tetraphenylporphyrins (H2ATPP) and the corresponding lutetium(III) complexes have been studied computationally at the density functional theory (DFT) and second-order algebraic diagrammatic construction (ADC(2)) levels using triple-ξ basis sets augmented with polarization functions. The molecular structures were optimized using Becke's three-parameter hybrid functional (B3LYP). The electronic excitation spectra in the range of 400–700 nm were calculated using the ADC(2) and the linear-response time-dependent DFT methods. The calculated spectra are compared to those measured in ethanol solution. The calculated excitation energies agree well with those deduced from the experimental spectra. The excitation energies for the Qx band calculated at the B3LYP and ADC(2) level are 0.20-0.25 eV larger than the experimental values. The excitation energies for the Qy band calculated at the B3LYP level are 0.10-0.20 eV smaller than the ADC(2) ones and are thus in good agreement with experiment. The calculated excitation energies corresponding to the Bx and By bands are 0.10-0.30 eV larger than the experimental values. The excitation energies of the Bx and By bands calculated at the B3LYP level are in somewhat better agreement with experiment than the ADC(2) ones. The calculated and measured band strengths largely agree.

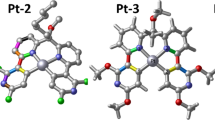
The ground-state molecular structures of H2TPP-EDTA, H2ATPP-DTPA, H2ATPPLuEDTA and H2ATPP-LuDTPA optimized at the B3LYP/TZVP level of theory


Similar content being viewed by others


References
Winpenny REP (1998) The structures and magnetic properties of complexes containing 3d- and 4f-metals. Chem Soc Rev 27:447–452
Send R, Sundholm D (2007) Coupled-cluster studies of the lowest excited states of the 11-cis-retinal chromophore. Phys Chem Chem Phys 9:2862–2867
Köhn A, Hättig C (2004) On the nature of the low-lying singlet states of 4-(dimethyl)aminobenzonitrile. J Am Chem Soc 126:7399–7410
Lehtonen O, Sundholm D (2006) Coupled-cluster studies of the electronic excitation spectra of silanes. J Chem Phys 125:144314
Sobolewski AL, Domcke W, Hättig C (2006) Photophysics of organic photostabilizers. Ab initio study of the excited-state deactivation mechanisms of 2-(2'-hydroxyphenyl)benzotriazole. J Phys Chem A 110:6301–6306
Hellweg A, Hättig C, Merke I, Stahl W (2006) Microwave and theoretical investigation of the internal rotation in m-cresol. J Chem Phys 124:204305
Schirmer J (1982) Beyond the random-phase approximation: a new approximation scheme for the polarization propagator. Phys Rev A 26:2395–2416
Trofimov AB, Schirmer (1995) An efficient polarization propagator approach to valence electron excitation spectra. J J Phys B 28:2299–2325
Hättig C (2005) Structure optimizations for excited states with correlated second-order methods: CC2 and ADC(2). Adv Quantum Chem 50:37–60
Bauernschmitt R, Ahlrichs R (1996) Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem Phys Lett 256:454–464
Furche F, Ahlrichs R (2002) Adiabatic time-dependent density functional methods for excited state properties. J Chem Phys 117:7433–7447
Hada M, Yokono H, Nakatsuji H (1987) Frozen core and virtual orbitals in the MCSCF theory. Chem Phys Lett 141:339–345
Adamowicz L, Bartlett RJ *1987) Optimized virtual orbital space for high-level correlated calculations. J Chem Phys 86:6314–6324
Sosa C, Geertsen J, Trucks GW, Bartlett RJ (1989) Selection of the reduced virtual space for correlated calculations. An application to the energy and dipole moment of Water. Chem Phys Lett 159:148–154
Taube AG, Bartlett RJ (2005) Frozen Natural orbitals: systematic basis set truncation for coupled cluster theory. Collect Czech Chem Commun 70:837–850
Neogrady P, Pitonak M, Urban M (2005) Optimized virtual orbitals for correlated calculations: an alternative approach. Mol Phys 103:2141–2157
Köhn A, Olsen J (2006) Coupled-cluster with active space selected higher amplitudes: performance of seminatural orbitals for ground and excited state calculations. J Chem Phys 125:174110
Piacenza M, Della Sala F, Fabiano E, Maiolo T, Gigli (2007) Torsional effects on excitation energies of thiophene derivatives induced by -substituents: comparison between time-dependent density functional theory and approximated coupled cluster approaches. G J Comput Chem 29:451–457
Send R, Kaila VRI, Sundholm D (2011) Reduction of the virtual space for coupled-cluster excitation energies of large molecules and embedded systems. J Chem Phys 134:214114
Pabst M, Sundholm D, Köhn A (2012) Ab initio studies of triplet-state properties for organic semiconductor molecules. J Phys Chem C (submitted)
Semenishin N, Rusakova N, Mazepa A, Korovin Y (2009) Synthesis of ditopic porphyrins and lanthanide complexes on their basis: luminescent features. Macroheterocycles 2(1):57–59
Gunes S, Neugebauer H, Sariciftci NS (2007) Conjugated polymer-based organic solar cells. Chem Rev 107:1324–1338
Evans R, Douglas P (2009) Design and color response of colorimetric multilumophore oxygen sensors. Appl Mater Interfaces 1:1023–1030
Evans R, Douglas P, Winscom C (2006) Coordination complexes exhibiting room-temperature phosphorescence: evaluation of their suitability as triplet emitters in organic light emitting diodes. Coord Chem Rev 250:2093–2126
Ermolina EG, Kuznetsova RT, Gadirov RM, Maier GV, Semenishin NN, Rusakova NV, Korovin YV (2010) Luminescence of the free bases of chelatee substituted tetraphenylporphyrin derivatives and their complexes with lutetium. High Energy Chem 44:387–424
Valiev RR, Ermolina EG, KaluginaYN KRT, Cherepanov VN (2012) Electronic absorption spectrum of monoamine tetraphenylporphyrin with the complexon of ethylenediaminetetraacetic acid as substitute. Spectrochim Acta A. doi:10.1016/j.saa.2011.10.075
Valiev RR, Ermolina EG, Kuznetsova, RT, Cherepanov VN (2012) Electronic absorption spectrum of monoamine tetraphenylporphyrin with the complexon of diethylentriamenepentaacetatic acid as substitute. Russian J Phys 54:1160–1165
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Ahlrichs R, Bär M, Häser M, Horn H, Kölmel C (1989) Electronic structure calculations on workstation computers: the program system TURBOMOLE. Chem Phys Lett 162:165–169, current version: see http://www.turbomole.com.
Schäfer A, Huber C, Ahlrichs R (1994) Fully optimized contracted gaussian-basis sets of triple zeta valence quality for atoms Li to Kr. J Chem Phys 100:5829–5835
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys Chem Chem Phys 7:3297–3305
Hättig C, Köhn A (2002) Transition moments and excited-state first-order properties in the coupled-cluster model CC2 using the resolution-of-the-identity approximation. J Chem Phys 117:6939–6952
Hättig C, Weigend FJ (2000) CC2 excitation energy calculations on large molecules using the resolution of the identity approximation. Chem Phys 113:5154–5161
Ladd MFC, Povey DC (1973) Crystallographic and spectroscopic studies on ethylenediaminetetraacetic acid (edta). J Chem Cryst 3:15–23
Shkolnikova LM, Polyanchuk GV, Dyatlova NM, Polyakova IA (1984) An X-ray study of organic ligands of complexon type. J Struct Chem 25:445–448
Aime S, Benetolloa F, Bombieri G, Colla S, Fasano M, Paoletti S (1997) Non-ionic Ln(III) chelates as MRI contrast agents: synthesis, characterisation and 1H NMR relaxometric investigations of bis(benzylamide)diethylenetriaminepentaacetic acid Lu(III) and Gd(III) complexes. Inorg Chim Acta 254:63–70
Silvers SJ, Tulinsky A (1967) The crystal and molecular structure of triclinic tetraphenylporphyrin. J Am Chem Soc 89:3331–3337
Gassman PG, Ghosh A, Almlöf J (1992) Electronic effects of peripheral substituents in porphyrins: x-ray photoelectron spectroscopy and ab initio self-consistent field calculations. J Am Chem Soc 114:9990–10000
McGlynn SP, Azumi T, Kinoshita M (1969) Molecular spectroscopy of the triplet state.Prentice-Hall Englewood Cliffs, NJ
Solov’ev KN, Gurinovich GP, Sevchenko AN (1963) The spectroscopy of the porphyrins. Sov Phys Usp 6:67–105
Baerends EJ, Ricciardi G, Rosa A, van Gisbergen SJA (2002) A DFT/TDDFT interpretation of the ground and excited states of porphyrin and porphyrazine complexes. Coord Chem Rev 2002:230
Acknowledgments
This work has been supported by the Academy of Finland through its Centers of Excellence Program 2006–2011. The calculations were performed at the CSC-IT Center for Science, Finland.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 914 kb)
Rights and permissions
About this article
Cite this article
Valiev, R.R., Ermolina, E.G., Kuznetsova, R.T. et al. Computational and experimental studies of the electronic excitation spectra of EDTA and DTPA substituted tetraphenylporphyrins and their Lu complexes. J Mol Model 19, 4631–4637 (2013). https://doi.org/10.1007/s00894-012-1400-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-012-1400-9