
Abstract
Calpainopathy is mainly characterized by symmetric and progressive weakness of proximal muscles. Several reports showed that the most common LGMD subtype is LGMDR1 or calpainopathy, which had previously been defined as LGMD2A. Until now, more than 500 likely pathogenic/pathogenic variants in the CAPN3 gene have been reported. However, a clear genotype–phenotype association had not yet been established and this causes major difficulties in predicting the prognosis in asymptomatic patients and in providing genetic counseling for prenatal diagnosis. In this report, we aimed to add new data to the literature by evaluating 37 patients with likely pathogenic/pathogenic variants for the detected variants’ nature, patients’ phenotypes, and histopathological features. As a result, the general clinical presentation of the 23 different variants was presented, the high frequency of NM_000070.3:c.550delA mutation in Exon 4 was discussed, and some novel genotype–phenotype associations were suggested. We have underlined that calpainopathy can be misdiagnosed with inflammatory myopathies histopathologically. We have also emphasized that, in young or adult patients with mild to moderate proximal muscle weakness and elevated CK levels, calpainopathy should be the first suspected diagnosis.


Similar content being viewed by others

References
Özyilmaz B et al (2019) Impact of next-generation sequencing panels in the evaluation of limb-girdle muscular dystrophies. Ann Hum Genet 83(5):331–347
Richard I et al (1995) Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 81(1):27–40
Murphy AP, Straub V (2015) The classification, natural history and treatment of the limb girdle muscular dystrophies. J Neuromuscul Dis 2(s2):S7–S19
Mohassel P, Bönnemann CG (2015) Limb-girdle muscular dystrophies. Neuromuscular disorders of infancy, childhood, and adolescence. Elsevier, pp 635–666
Pathak P et al (2020) Mutational spectrum of CAPN3 with genotype-phenotype correlations in limb girdle muscular dystrophy type 2A/R1 (LGMD2A/LGMDR1) patients in India. J Neuromuscul Dis, (Preprint): p. 1–12
Richard I et al (2016) Natural history of LGMD 2A for delineating outcome measures in clinical trials. Ann Clin Transl Neurol 3(4):248–265
Gallardo E, Saenz A, Illa I (2011) Limb-girdle muscular dystrophy 2A. Handb Clin Neurol 101:97–110
Ojima K et al (2011) Non-proteolytic functions of calpain-3 in sarcoplasmic reticulum in skeletal muscles. J Mol Biol 407(3):439–449
Aartsma-Rus A et al (2006) Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve: Official Journal of the American Association of Electrodiagnostic Medicine 34(2):135–144
Richards S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–423
Blázquez L et al (2008) Characterization of novel CAPN3 isoforms in white blood cells: an alternative approach for limb-girdle muscular dystrophy 2A diagnosis. Neurogenetics 9(3):173–182
Nilsson MI et al (2014) Redox state and mitochondrial respiratory chain function in skeletal muscle of LGMD2A patients. PLoS One 9(7):e102549
Nallamilli BRR et al (2018) Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol 5(12):1574–1587
Yİş U et al (2018) Childhood onset limb-girdle muscular dystrophies in the Aegean part of Turkey. Acta Myologica. 37(3):210
Stehlíková K et al (2014) Autosomal recessive limb-girdle muscular dystrophies in the Czech Republic. BMC Neurol 14(1):1–9
Dinçer P et al (2000) A cross section of autosomal recessive limb-girdle muscular dystrophies in 38 families. J Med Genet 37(5):361–367
Zhong H et al (2020) Molecular landscape of CAPN3 mutations in limb-girdle muscular dystrophy type R1: from a Chinese multicentre analysis to a worldwide perspective. J Med Genet
Saenz A et al (2005) LGMD2A: genotype–phenotype correlations based on a large mutational survey on the calpain 3 gene. Brain 128(4):732–742
Topaloǧlu H et al (1997) Calpain-3 deficiency causes a mild muscular dystrophy in childhood. Neuropediatrics 28(04):212–216
Balci B et al (2006) Calpain-3 mutations in Turkey. Eur J Pediatr 165(5):293–298
Dorobek M et al (2015) The frequency of c. 550delA mutation of the CANP3 gene in the Polish LGMD2A population. Genet Test Mol Biomark 19(11):637–640
Fanin M, Angelini C (2015) Protein and genetic diagnosis of limb girdle muscular dystrophy type 2A: the yield and the pitfalls. Muscle Nerve 52(2):163–173
Inashkina I et al (2016) Robust genotyping tool for autosomal recessive type of limb-girdle muscular dystrophies. BMC Musculoskelet Disord 17(1):1–6
Vissing J et al (2020) A single c. 1715G> C calpain 3 gene variant causes dominant calpainopathy with loss of calpain 3 expression and activity. Human Mutat 41(9):1507–1513
Zhang C et al (2021) Compound heterozygous CAPN3 variants identified in a family with limb-girdle muscular dystrophy recessive 1. Mol Med Rep 23(6):1–8
Piluso G et al (2005) Extensive scanning of the calpain-3 gene broadens the spectrum of LGMD2A phenotypes. J Med Genet 42(9):686–693
Fanin M et al (2005) The frequency of limb girdle muscular dystrophy 2A in northeastern Italy. Neuromuscul Disord 15(3):218–224
Alharbi N et al (2021) Clinical and genetic features of calpainopathies in Saudi Arabia–a descriptive cross-sectional study. Eur Rev Med Pharmacol Sci 25(15):4941–4952
Anderson LV et al (2000) Secondary reduction in calpain 3 expression in patients with limb girdle muscular dystrophy type 2B and Miyoshi myopathy (primary dysferlinopathies). Neuromuscul Disord 10(8):553–559
Diniz G et al (2012) A regional panorama of dysferlinopathies. Turk Patoloji Derg 28(3):259–265
Han R (2011) Muscle membrane repair and inflammatory attack in dysferlinopathy. Skelet Muscle 1(1):1–8
Author information
Authors and Affiliations
Contributions
All of the authors contributed to the design and implementation of the research, to the analysis of the results, and the writing of the manuscript.
Corresponding author
Ethics declarations
Ethical publication statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Ethics approval and consent to participate
The study was not submitted to a research ethics committee. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975 (in its most recently amended version). Informed consent was obtained from all patients included in the study. Additional informed consent was obtained from all patients for whom identifying information is included in this article. All institutional and national guidelines for the care and use of laboratory animals were followed. This article does not contain any studies with human or animal subjects.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Remarks
• In young or adult patients with mild to moderate proximal muscle weakness and elevated CK levels, calpainopathy should be the first suspected diagnosis.
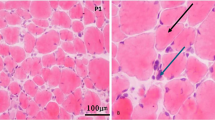
• Histopathology of calpainopathy may be accompanied by inflammatory findings. Thus, we suggest that calpainopathies should be kept in mind in both differential diagnosis and molecular genetic test planning in patients with suspected inflammatory myopathy.
• Among 359 patients, we have evaluated for the diagnosis of muscular dystrophy; 37 (10.3%) had one or more pathogenic/likely pathogenic variants in the CAPN3 gene. Three of the variants were not found in databases and literature; thus, they were interpreted as novel pathogenic variants.
• The NM_000070.3:c.550delA variant at exon 4, was the most frequent variant (5 homozygous patients, 10 alleles) in this report. c.550delA variant is usually associated with moderate phenotype progressing to loss of ambulation in later decades.
• The NM_000070.3:c.597_611delGTTCTGGAGTGCTCT variant was recently associated with autosomal dominant inheritance; however, with the data we acquired in this study, we suggest that it may be associated with both autosomal recessive and autosomal dominant forms of calpainopathy.
Rights and permissions
About this article

Cite this article
Ozyilmaz, B., Kirbiyik, O., Ozdemir, T.R. et al. Experiences in the molecular genetic and histopathological evaluation of calpainopathies. Neurogenetics 23, 103–114 (2022). https://doi.org/10.1007/s10048-022-00687-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-022-00687-4