
Abstract
Background
Type I gastric neuroendocrine tumors (gNETs) arise from hypergastrinemia in patients with autoimmune chronic atrophic gastritis. According to the classical model, the gastric H+/K+ ATPase was the causative autoantigen recognized by CD4+ T cells in chronic autoimmune scenario that secretes IL-17 and correlates with parietal cell (PC) atrophy, which drives to gastric achlorhydria and increases the risk for gastric neoplasms. However, the mechanism by which the inflammatory response correlates with PC atrophy is not clearly defined.
Methods
Recently, we found that the ATP4Ap.R703C mutation impaired PC function and gastric acidification, which drove familial gNET. Our group constructed a knock-in mouse model for the ATP4A mutation, which has served us to better understand the relation between impaired capability to export protons across the plasma membrane of PCs and tumor progression.
Results
The ATP4Ap.R703C mutation drives gastric achlorhydria, but also deregulates the acid–base balance within PCs, affecting mitochondrial biogenesis. Mitochondrial malfunction activates ROS signaling, which triggers caspase-3-mediated apoptosis of parietal cells. In addition, when gastric euchlorhydria was restored, mitochondrial function is recovered. Infection by H. pylori promotes destabilization of the mitochondria of the PCs by a mechanism similar to that described for APT4Ap.R703C carriers.
Conclusions
A genetic origin that drives mitochondria alteration would initiate the gastric chronic inflammation instead of the classical IL-17 secretion-mediated mechanism explanation. Gastric euchlorhydria restoration is suggested to be indicated for mitochondrial recover. Our results open a new window to understand gastric neoplasms formation but also the inflammatory mechanisms and autoimmune disorders conducted by genetic origin that composes a premalignant scenario.
Similar content being viewed by others

Introduction
Gastric acidification is achieved through the H+/K+ proton pump (ATP4A) of the parietal cells (PCs), which release H+ to form gastric acid (HCl) in the gastric lumen [1]. PCs are positively regulated by enterochromaffin-like (ECL) cells after binding of gastrin to the CCKB receptor (see Ref. [2] for a review). PC and ECL cells are located in the base of the oxyntic glands, which differentiate from neck mucosa cells (NMCs) in the upper part of the gland (isthmus) [3]. Proliferation and migration of differentiated NMCs are coordinated by the acidity of the stomach and different growth factors activated by gastrin.
Type I gastric neuroendocrine tumors (gNETs) have a low proliferation rate (based on Ki67 staining) and arise in patients with autoimmune chronic atrophic gastritis [4]. gNETs are diagnosed by positive staining for the biomarkers chromogranin A (chr), synaptophysin, and somatostatin receptor 2 (SSTR2) [5]. Tumor progression is classically described to arise from the hypergastrinemia that caused the chronic gastritis, followed by neoplasia of ECL cells; it involves destruction of PCs (atrophy) and correlates with gastric achlorhydria [6]. However, we recently described different families affected with gNETs and autoimmune chronic atrophic gastritis, which was explained by mutations in the ATP4A and PTH1R genes, both involved in gastric acid regulation [7, 8]. A knock-in mouse model (KI/KI) with one of the mutations found in homozygosis in the human ATP4A gene (p.R703C) also lacked the capability to transport protons across the plasma membrane of PCs, impairing the acidification of the stomach. However, 1-year-old KI/KI mice (KI/KI1) only recapitulated premalignant conditions such as inflammation, atrophic gastritis, and metaplasia [9]. Even though none of the mouse models for genes involved in chlorhydria/gastrin regulation recapitulate gastric dysplasia (see Ref. [10] for a review), our KI/KI model allowed us to assess the relationship of proton pump malfunction and the lack of gastric chlorhydria with hypergastrinemia and gNET progression. In addition, treatment of KI/KI mice with acidified water in the diet both prevented and reverted the development of the premalignant phenotype, demonstrating that achlorhydria was the primary cause of tumorigenesis in families affected with mutations in the ATP4A gene [9].
Besides the genetic origin (mutations in the ATP4A gene), other risk factors have been described for gastric neoplasms severe dysplasia, gastric cancer [11], and gNETs [12]. Human autoimmune gastritis is been described to arise from IL-17-depending innate autoimmune response mediated by CDH+ T cells [13], which recognize H+/K+ ATPase (ATP4A) (anti-parietal cells antibodies) [14], and activation of the NLRP3 inflammasome-reactive oxygen species (ROS) pathway [15]. NLRP inflammasome is a multi-protein intracellular pattern recognition receptor complex that facilitates the cleavage of the inflammatory caspases and secretion of the pro-inflammatory cytokines which, in turn, triggers an immune response [16]. Recently, using a mouse model that expresses a transgenic T-cell-receptor specific for the autoantigen H+/K+ ATP4A (TxA23) [17], a correlation between the grade of gastric atrophy and the pro-inflammatory cytokine IL-17 was described [18]. Caspase-3-dependent apoptosis of parietal cells was observed in vitro and in organoids derived from the TxA23 model [18].
On the other hand, H. pylori mimics PC antigens in human autoimmune gastritis, thus preventing the adaptive immune response and reducing gastric acidity [19]. In addition, secretion of IL-17 has also been described in H. pylori infection. Treatment of H. pylori-infected mice with an anti-IL-17 antibody significantly reduced the H. pylori burden and inflammation of the stomach [20].
Both risk factors (autoimmune response and H. pylori infection) involve gastritis that progresses to chronic inflammation, PC atrophy, neck mucous cell (NMC) hyperplasia, metaplasia, and eventually intraepithelial dysplasia [21]. However, the mechanism by which the inflammatory response correlates with IL-17 secretion and PC atrophy is not fully understood [12]. In the present study, we investigated the genetic inactivation of ATP4A and chronic gastritis in our KI/KI mouse model to better understand their correlation with the autoimmune response and neoplasia progression in human patients with mutations affecting gastric euchlorhydria.
Materials and methods
Mice
A previously published [9] knock-in mouse model (KI/KI) for the human mutation ATP4Ap.R703C was used in this study. All animal procedures were previously approved by CNIO (Register number: IACUC.043–2014) and the Institutional Animal Ethics Committee (Register number: PROEX292/14). The mouse model is registered in the MGI database (Atp4atm1.1Ocalv, MGI: 6,118,153, URL: https://www.informatics.jax.org/allele/MGI:6118153). Sample collecting was performed according ARRIVE guidelines.
Samples
DNA and RNA samples were collected from formalin-fixed, paraffin-embedded (FFPE) gastric tissues from three wild-type (wt) mice and three 2-year-old KI/KI mice (KI/KI2). Samples from two different regions of the stomach were studied: positive for the anti-chromogranin A antibody (chr+) and negative for the anti-chromogranin A antibody (chr−), which correspond to the premalignant conditions observed in the 1-year-old KI/KI mice (KI/KI1). DNA and RNA samples from gastric tissue of KI/KI2 mice from prevention and reversion studies were also collected [9]. No gender-related differences were previously found, and therefore, no biological replicas regarding male and female mice were considered at this point.
DNA and RNA samples of FFPE gastric tissue patients described in Calvete et al. [7] (homozygous for the ATP4Ap.R703C mutation) and Calvete et al. [8] (heterozygous for the ATP4Ap.Q680L mutation) were also collected. Control DNA and RNA (Wt) were obtained from FFPE gastric tissue from two stomach reduction surgery of patients without gastric disease. IHC performed in gNET patients were replicated in a series of six patients infected with H. pylori. DNA and RNA extraction was performed from FFPE tissues using the Maxwell® RSC DNA FFPE Kit following the manufacturer’s instructions. All samples were collected from the fundus/corpus area of the stomach, where the oxyntic glands are located.
mtDNA/nDNA ratio analysis
mtDNA/nDNA ratio was calculated comparing the expression of mitochondrial-encoded genes versus nuclear-encoded genes. In mice, mt-co1 and tfam were used as mitochondrial and nuclear-encoded genes respectively. In humans, the same analysis was performed using the MT-RNR2 and GAPDH genes. Expression of Tbp and 36B4 genes was used to normalize the obtained results for mouse and human samples, respectively.
RNA samples and quantitative RT-PCR
Five hundred nanograms of total RNA from each sample were reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) in a final volume of 20 µL using a standard protocol. The concentration and purity of isolated RNA were assessed by NanoDrop™ 2000 and 2000c Spectrophotometers (Thermo Fisher Scientific). All primers were designed with Primer3 software (v. 0.4.0). The sequences of the primers are listed in Supplementary Table 1. Quantitative RT-PCR (qRT-PCR) using the GoTaq® qPCR Master Mix (Promega) was performed to evaluate gene expression. Samples were amplified using the standard cycling conditions. All qRT-PCR reactions were performed in triplicate. Tbp and GAPDH were used as internal controls in mice and humans, respectively. The analysis was performed using QuantStudio™ Real-Time PCR Software v1.3. The 2−ΔCt method was applied to calculate the relative expression of genes for each sample.
Immunohistochemical studies
Blocks were cut into 5-μm-thick sections and stained with Hematoxylin and Eosin (H&E) for light microscopy examination. Details of antibodies used in mouse and human FFPE are shown in Supplementary Tables 2 and 3, respectively.
Electron microscopy
Electron microscopy was performed on gastric tissue of a KI/KI1 mouse to evaluate tumor progression. One square millimeter slices of stomachs from a wt and a KI/KI1 mouse were fixed overnight with paraformaldehyde (4%) + glutaraldehyde (2%) in Hepes buffer (0.4 M) at 4 °C Fixer solution was removed the next day and samples were washed with 0.4 M Hepes buffer. Sections of 1 µm thick were stained with toluidine blue and examined to select areas of interest. Selected sections were embedded in Spurr’s resin and ultrathin sections were cut using a microtome for detailed examination by electron microscopy.
Results
Evaluation of 2-year-old mice
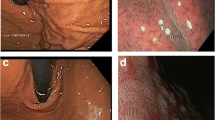
One-year-old KI/KI mice (KI/KI1) recapitulated most of the symptoms seen in human pathology, including hypergastrinemia caused by gastric achlorhydria. However, only premalignant conditions, chronic gastritis and mucous metaplasia, were observed, whereas increased chromogranin A (chr) staining was not observed. We monitored the KI/KI mice a year later, and observed tumor development at 2 years (KI/KI2) (Fig. 1a). Focal dysplastic positive staining for chr and SSTR2 and low Ki67 staining was observed in these mice (Fig. 1b). Further studies were performed on chr positive (+) and chr negative (−) areas. Chr+ samples correspond to KI/KI2 mice positive for tumor biomarkers, while chr− samples correspond to premalignant conditions as observed in the KI/KI1 mice. Delayed positive chr staining suggested a slower progression of gNETs in mice compared to human patients carrying the same mutation, which may indicate with a different response to hypergastrinemia conditions in mice and humans.

Gastric histopathology of the KI/KI mice. a Macroscopic image of representative wt and KI/KI2 stomachs. Scale bar (shown in white): 1 cm. b Representative H&E and IHC staining of a KI/KI2 mouse stomach. Muscular layer and gastric lumen are indicated with white and black arrows, respectively. Left panel; H&E staining shows not only the premalignant condition with high number of epidermoid cysts and vacuolization of gastric mucosa cells observed in the 1-year-old KI/KI1 mice, but also dysplastic cell masses (black arrowhead). Right panel: IHC with anti-ChrA, SSTR2 and Ki67 antibodies of a representative KI/KI2 stomach. c IHC with anti-P-S6 antibody. Left panels: staining in human gastric tissue as indicated. P-S6 expression is increased by gastrin mainly in ECL cells (base of the glands) in the stomach of a healthy subject (wt). IHC staining in normal (N) tissue of a gNET ATP4Ap.R703C patient. Increased staining correlates with increased activation of the CCKBR receptor in a massive response to hypergastrinemia. Right panels: staining in mouse gastric tissue as indicated. IHC with anti-P-S6 antibody also stains the ECL cells at the base of the glands and the isthmus region. Increased expression is not observed in the KI/KI1 mouse compared to wt stomach, but increased staining is seen in the KI/KI2 mouse. d Electron microscopy (EM) of a wt mouse gastric neck mucosa section. Colored schematic representation of the cell types is shown in the cell type’s legend. PC cells show prominent cytoplasmic tubulovesicles and intracellular canaliculi containing short microvilli along their apical surface and numerous mitochondria. Scale bar (shown in red): 1000 µm. e Left panel: EM of a KI/KI1 mouse neck mucosa section. ECL and NMC have a reduced number of granules and are heavily vesiculated and hyperplasied, respectively. Right panel: detail of a representative PC with an open-wide canaliculus (asterisk), abnormal mitochondria (white arrowheads), and glycogen granules (white arrows). Scale bar (shown in white): 1 μm. NMC neck mucosa cells (proliferating), ECL enterochromaffin cells, PC parietal cells, ZC zymogenic cells (chief cells)
Different response to hypergastrinemia in humans and mice
To evaluate the delayed gNET formation in mice, we explored the role of hypergastrinemia in gastric mucosa proliferation. Gastrin activates the gastrin receptor (CCKBR) in ECL cells to regulate proliferation, development, and homeostasis of oxyntic glands through MAPK and PI3K/mTOR pathways. Activation of MAPK and PI3K/mTOR pathways was tested using antibodies against activated (phosphorylated) forms of P38 and S6, respectively. First, non-tumor tissues from a human patient homozygous for the ATP4Ap.R703C mutation and from KI/KI1 mice were compared with healthy stomachs (normal tissue from non-affected human and mouse). Slight differences regarding the P-P38 staining were found between healthy and ATP4Ap.R703C mutation carriers in humans that were not observed in mice (Supplementary Fig. 1). However, different P-S6 staining patterns were observed; in humans, staining with anti P-S6 was predominantly located in the basal region of the oxyntic glands (ECL cells) in Wt tissue, while normal (N) tissue of gNET patients with the ATP4Ap.R703C mutation showed overexpression of this phosphorylated protein in the entire gastric wall (Fig. 1c). In wt mice, this phosphorylated protein was also highly expressed in the basal region of the oxyntic glands and in the isthmus (proliferation area). However, the KI/KI1 mouse model did not overexpress this phosphorylated protein. Finally, a slightly increased P-S6 staining signal was encountered in the KI/KI2 mice (Fig. 1c), showing a delay in the hypergastrinemia response. The P-S6 staining patterns suggest a different proliferation activity between mice and humans.
Cellular integrity in the achlorhydric mouse model (KI/KI)
Cellular integrity and morphology of KI/KI1 mouse stomachs were evaluated with electron microscopy (EM) to better understand the effect of achlorhydria during tumor progression. The oxyntic glands of wt mouse stomachs were first evaluated. PCs have prominent cytoplasmic tubulovesicles, numerous mitochondria, and intracellular canaliculi that contain short microvilli along their apical surface. Enterochromaffin-like cells and chief (zymogenic) cells contain abundant vesicles and prominent rough endoplasmic reticulum (RER), respectively. NMC cells, which differentiate while migrating to the base of the oxyntic glands, were also commonly observed (Fig. 1d).
EM revealed no loss of any cell type in KI/KI1 mice (Fig. 1e). However, evident morphology changes and certain disordered cytoplasms were observed. The RER of chief cells was unstructured and less abundant, and ECL cells had a lower amount of secreting granules. Finally, open-wide canaliculi and fewer and shorter microvilli instead of normal tubulovesicles were found in PCs. Abnormal mitochondria and abundant granules of glycogen were observed in the cytoplasm.
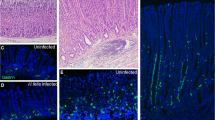
We hypothesized that the lack of H+ export would alter the internal acid–base balance of PCs, which might alter mitochondrial function resulting in the observed abnormal mitochondria. We, therefore, tested the acid–base balance in the KI/KI2 mouse model with anti-carbonic anhydrase IX (CA IX) antibodies; CA IX is associated with the cell membrane and directly participates in the acid–base balance in PCs. Decreased staining was observed in chr+ tissue compared to wt stomach and chr− tissues (Fig. 2a).

Evaluation of parietal cells and mitochondrial function in wt and KI/KI2 mice. a Representative IHC staining with anti-pyruvate carboxylase (PC) and anti-carbonic anhydrase IX (CA IX) antibodies in wt (upper panel) and KI/KI2 mice (chr+ and chr−) (lower panel); relative mt-Cytb expression in gastric tissues from 2-year-old mice is also shown. Detailed (large scale) images are shown for KI/KI2 gastric tissues. b Mitochondrial biogenesis: mtDNA/nDNA ratio and relative Polg and Tfam expression in wt and KI/KI2 (chr+ and chr−) gastric tissues from 2-year-old mice. c Oxidative stress response: relative expression of Sod1, Sod2, and catalase in wt and KI/KI2 (chr+ and chr−) gastric tissues from 2-year-old mice. d Evaluation of mitochondria in KI/KI2 mice prevention (prev) and reversion (rev) models (restoration of euchlorhydria)
Mitochondrial activity, biogenesis, and ROS activation of the mitochondrial damage response
Mitochondrial respiration was further evaluated. No differences were observed in staining with anti-DLST and anti-SDHA antibodies, which visualize intermediate metabolites from the Krebs cycle and the Complex II from OXPHOS (mitochondrial respiration), respectively (Supplementary Fig. 1). However, immunohistochemistry (IHC) with anti-pyruvate carboxylase (an enzyme of the Krebs cycle) revealed a decreased staining in chr+ KI/KI2 gastric tissue compared to wt stomach (Fig. 2a). Oxidative phosphorylation was also evaluated by measuring gene expression levels of cytochrome B (CytB; a subunit of the respiratory chain complex III). Down-regulation of gene expression was observed in both chr− and chr+ tissues compared to wt tissue (Fig. 2a).
Mitochondrial biogenesis was evaluated by assessing the levels of mitochondrial DNA polymerase γ (Polg) and the transcription factor Tfam, the main regulators of mitochondrial biogenesis. A marked up-regulation of Polg and Tfam was observed in chr+ tissue compared to chr− and wt gastric tissues, even though we observed a reduction of mtDNA levels in the chr+ tissue. Tfam might be increased to compensate low mtDNA levels (Fig. 2b).
To validate whether mitochondrial dysfunction activated the mitochondrial damage response, we determined the gene expression levels of the main enzymes activated by oxidative damage caused by reactive oxygen species (ROS). We found a notably increased expression of superoxide dismutase 1 (SOD1) and superoxide dismutase 2 (SOD2) compared to wt tissue. Of note, no differences were found in catalase gene expression of chr+ tissue compared to wt stomach (Fig. 2c). Since catalase only has a cytosolic function, this result allowed us to localize the damage to the mitochondria, rather than to the cytoplasm of the PCs.
Mitochondrial function in KI/KI mice with restored euchlorhydria conditions
Finally, to correlate mitochondrial damage and activated oxidative stress response (ROS) with a genetic origin (ATP4Ap.R703C mutation), we explored the mitochondrial activity in KI/KI2 mice treated with acidified water from birth (prevention model) or during adulthood (reversion model), as previously described [9]. Administration of HCl-water from birth (prevention) restored CytB expression levels and raised mtDNA content. Moreover, the treatment decreased Polg and Sod2 expression levels, although Sod1 expression was not affected, while Tfam levels increased compared to wt and untreated KI/KI mice (Fig. 2d). Similarly, administration of HCl-acidified water during adulthood (reversion model) increased CytB and mtDNA copy number and lowered Polg and Sod2 expression. Contrary to the prevention model, in the reverted phenotype, Tfam expression reached wt levels, while Sod1 expression was markedly reduced (Fig. 2d). Therefore, KI/KI2 mice treated with acidified water in the diet showed partial or complete restoration of the mitochondrial phenotype, demonstrating that achlorhydria was the main cause of mitochondrial damage.
Mitochondrial damage in KI/KI mice induces apoptosis and inflammation
The increase of ROS in the cell activates apoptosis and inflammation. In addition, recent studies have described that apoptosis drives PC atrophy in spasmolytic polypeptide-expressing metaplasia (SPEM), which is a preneoplastic process [22]. We therefore performed IHC studies to evaluate the levels of p21 (cell cycle arrest under damage conditions) and caspase-3 (activates apoptosis) in the KI/KI2 mouse model. Increased staining was found with both anti-p21 and anti-caspase-3 antibodies in chr+ tissue compared to the chr− and wt tissue (Fig. 3a). Furthermore, we observed an increased expression of the IL1b gene (a marker of inflammation), and an increased ICAM1 gene expression; ICAM1 is required for the invasion of the gastric mucosa by mature T cells during the autoimmune response (Fig. 3b). However, IHC with an antibody against IL-17, the cytokine that mediates the autoimmune response in chronic atrophic gastritis, had no staining in chr− or chr+ tissue of the KI/KI2 mice (Fig. 3c).

Activation of apoptotic pathway and inflammation in KI/KI2 mice. a Representative IHC staining with anti-P21 and anti-caspase-3 antibodies in wt and KI/KI2 (chr+ and chr−) gastric tissues from 2-year-old mice. Detailed (large scale) images are shown for KI/KI2 gastric tissues. b RT-qPCR of Il-1b and Icam-1 mRNA isolated from wt and KI/KI2 (chr+ and chr−) FFPE tissues. c Representative IHC staining with anti-IL-17 antibody in gastric tissues from 2-year-old KI/KI2 (chr− and chr+ ) mice
Human ATP4Ap.R703C patients and H. pylori-affected patients exhibit mitochondrial damage, oxidative stress, and caspase-3-mediated apoptosis of parietal cells
To translate our results from the KI/KI2 mouse model to human patients, we first evaluated mitochondrial biogenesis and function in FFPE tissue from individuals affected with gNETs carrying the ATP4Ap.R703C mutation from a previously studied family [7]. mtDNA content and POLG and TFAM expression increased in tumor tissue (T) compared to non-tumor (N) stomach tissue of the patients (Fig. 4a). Furthermore, SOD1, SOD2, and catalase were markedly over-expressed in tumor tissue compared to non-tumor and control tissues (Fig. 4a). Interestingly, it was recently described that H. pylori infection involves up-regulation of POLG and TFAM to alter mitochondrial replication of the host cell [23]. We speculated that the inflammation progression induced by achlorhydria might be similar to that induced by H. pylori infection, and we, therefore, evaluated acid–base balance and caspase-3-mediated apoptosis in a gastric biopsy from a patient infected with H. pylori. IHC studies with anti-carbonic anhydrase IX antibodies showed a reduced expression of the protein in tumor tissues of the ATP4Ap.R703C patient and the H. pylori-affected patient compared to control tissue (Fig. 4b). Finally, IHC studies showed the increased expression of P21 and caspase-3 in ATP4Ap.R703C patients and H. pylori—infected tissues compared to control tissue. In addition, the same IHC studies were performed for patients carrying the ATP4Ap.Q680L mutation described in Calvete et al. [8]. The same observation was found in these individuals with a mutation in the ATP4A gene (Supplementary Fig. 2). Anti-IL-17 antibody staining was not found increased in the N tissue of ATP4Ap.R703C patients compared to normal stomach (Fig. 4c).

Evaluation of parietal cells and mitochondrial function in gNET human patients carrying the ATP4Ap.R703CC mutation. a Mitochondrial biogenesis (mtDNA/nDNA ratio, POLG, and TFAM) and oxidative stress response (SOD1, SOD2, and catalase expression) in gastric tissue from a healthy individual (Wt), compared to normal (N) and tumor (T) gastric tissue of a gNET patient carrying the ATP4Ap.R703CC mutation. b Representative IHC staining with anti-carbonic anhydrase IX (CA IX) antibody in gastric tissue from a healthy individual (Wt), in normal (N) and tumor (T) gastric tissues of a gNET patient carrying the ATP4Ap.R703CC mutation, and in gastric tissue of a patient with H. pylori (HP) infection. c Representative IHC staining with anti-p21, anti-caspase-3, and anti-IL-17 antibodies in gastric tissue from a healthy individual (wt), a gNET patient carrying the ATP4Ap.R703CC mutation (N and T gastric tissue) and gastric tissue of a patient with H. pylori (HP) infection. Black squares show detailed (larger scale) images
Discussion
In a previous study, we observed that 1-year-old KI/KI (KI/KI1) mice recapitulated the achlorhydria and hypergastrinemia scenario, but only the dysplastic premalignant conditions of gNETs [9]. In this work, we observed that the 2-year-old KI/KI mouse model (KI/KI2) developed positive staining for gNET type I diagnostic biomarkers (chr-A and SSTR2) and a low KI67 proliferation index (Fig. 1b). This is the first observation that the mouse model recapitulates gastric dysplasia regarding genes involved in the chlorhydria/gastrin regulation (see Ref. [10] for a review). In addition, our observations indicate a delayed tumor progression in mice compared to humans.
CCKBR (gastrin receptor) activates the proliferation pathway involved in oxyntic gland homeostasis. Our results suggest that increased anti-P-S6 staining in normal (N) tissue of gNET human patients corresponds to a massive response to gastrin, which increases cell proliferation and hyperplasia of ECL cells in an achlorhydria scenario. In the KI/KI mouse model, the lack of euchlorhydria also leads to hypergastrinemia. However, the gastrin receptor (CCKBR) did not massively activate proliferation as observed in human patients (Fig. 1c), which suggests a different response to high gastrin levels regarding proliferation activity.
Mitochondrial dysfunction
Electron microscopy revealed important changes in the morphology of PCs (Fig. 1). The presence of atypical open-wide canaliculi, abnormal mitochondrial morphology, and the accumulation of glycogen granules in the PCs of the KI/KI1 mice were also observed in the other knock-out mouse models for genes involved in gastric acid regulation, and correlate with a condition of achlorhydria [10, 24].
Our studies with anti-carbonic anhydrase IX antibody suggest that an impaired acid–base balance within the PC affects mitochondrial function and biogenesis (Fig. 2). On the other hand, the marked increase of superoxide dismutase (SOD) suggests activation of ROS-related oxidative damage signaling in the KI/KI mouse that takes place inside the mitochondria, since no differences were observed in catalase expression (cytoplasmic ROS increase catalase expression). Importantly, when the same evaluation was carried out in KI/KI2 mice treated with acidified water (prevention and reversion), mitochondrial activity and SOD levels were partially restored (Fig. 2d), which indicates a correlation between mitochondrial malfunction and damage activation. By restoring euchlorhydria, the acid–base balance in the PCs may be also restored, or at least the accumulation of H+ in the cytoplasm may be prevented, which we have shown to interfere with mitochondrial function.
Chr+ areas of KI/KI2 mouse stomachs showed increased expression of p21 (a marker of DNA damage and cell cycle arrest) and caspase-3 (an effector protein of the apoptotic pathway), which links the mitochondrial damage to apoptosis (Fig. 2d). This observation is in agreement with the low proliferation rate which we already observed (low Ki67 proliferation index). Apoptosis has been previously proposed as a mechanism of parietal cell atrophy that precedes SPEM [25].
In humans, a gNET patient carrying the ATP4Ap.R703C mutation also exhibited impaired oxidative phosphorylation activity, increased oxidative stress response, and increased mitochondrial replication that leads to activation of PC apoptosis, as observed in the chr+ KI/KI2 mouse model (Fig. 4). The same observation was found in the patients carrying the ATP4Ap.Q680L mutation (Supplementary Fig. 2). The loss of parietal cells has been described to increase proliferation of gastric stem cells and progenitor cells [26], which is in agreement with increased P-S6 expression (Fig. 1).
In summary, our results indicate that the lack of H+ export from PCs may alter the internal acid–base balance and mitochondrial function, probably through alteration of the favorable gradient between membranes that may interfere the function of porins and the ATP synthase (Fig. 5), which is in agreement with lack of activity of carbonic anhydrase (Fig. 2). The NLRP3 inflammasome is thought to sense the disturbance of cellular homeostasis including K+ efflux, Ca2+ signaling, and mitochondrial dysfunction rather than directly recognizing a common motif present in its activators [27]. In vitro studies have suggested that Ca2+mediated mitochondrial damage may activate the NLRP3 inflammasome [28]. Therefore, mitochondria membrane depolarization would trigger the ROS activation and the caspase-3-mediated apoptosis that lead to atrophy of PCs. Mitochondrial damage and ROS have been shown to be essential for NLRP inflammasome activation [29, 30].

Proton generation and transport in parietal cells. a Wild-type parietal cells. Proton pumps and channels generating favorable proton gradients are shown (green). Location of the different metabolic stages of mitochondrial respiration is also shown (red). Oxidative phosphorylation is a metabolic pathway within the inner membranes of the mitochondria that transports electrons (OXPHOS complex) to release H+ to the intermembrane space. The favorable proton gradient is used by ATP synthase to catalyze the formation of adenosine triphosphate. On the other hand, carbonic anhydrase participates in H+ production in the cell cytoplasm, which is released by ATP4A (proton pump) out of the cell to form HCl (gastric acid). b ATP4A-deficient cells. Proton pump malfunction prevents H+ from being released from the cell, which results in an alteration of the internal proton concentration of the parietal cell. Accumulation of H+ within the cell may alter the favorable gradients used by the mitochondria. Our results show that malfunction of the mitochondria correlates with impaired oxidative phosphorylation activity and activation of the oxidative stress response (ROS signaling), which induces parietal cell atrophy
Finally, IL-1b and Icam-1 (autoimmune response) were found increased in chr+ areas of the stomachs of KI/KI2 mice, which indicates that more cytokines may be involved in driving gastric diseases, as previously suggested [31]. NLRP inflammasome facilitates the secretion of IL-1b, which in turn triggers an immune response [16]. A correlation between mitochondrial damage, ROS activity and active IL-1b has been previously described [28]. Different expression levels of cytokines were also found in patients with different gastric neoplasms [32], including IL-17 [33]. Thus, the inflammation mechanism must be further evaluated to improve premature diagnosis and the identification of therapeutic targets. Indeed, the NLRP inflammasome has been suggested to be a promising therapeutic target for autoimmune diseases [34].
Genetic origin-mediated autoimmunity mechanism
According to the classical model, cytokines released by activated immune cells induce PC atrophy and gastric lesions [35, 36]. Specifically, IL-17 was identified as a critical cytokine secreted by CD4+ T helper 17 cells (Th17) and CD8+ T cells of the inflammatory microenvironment that induces ROS production, which triggers parietal cell death and autoimmune gastritis (Fig. 6a) [37]. Correlation between IL-17 secretion and the degree of parietal cell atrophy by inducing caspase-dependent cell death was observed in the TxA23 mouse model (autoreactive for ATP4A) [18]. However, why PCs express the IL-17 receptor in chronic gastritis was not explained.

Gastritis mechanisms. a IL-17-dependent autoimmunity. Inflammation mediates IL-17 cytokine release by activated immune cells (CD4) that results in PC atrophy and gastric lesions. b Genetic-origin-dependent gastritis. The ATP4Ap.R703C mutation drives gastric achlorhydria and impairs acid–base balance that results in mitochondrial malfunction and ROS activation of the mitochondrial damage response (red arrow), which drives PC atrophy and inflammation (green arrows). c H. pylori infection gastritis. H. pylori infection correlates with an increase of the gastric acid secretion from host (adaptive immunity to prevent infection) (red arrow). To prevent innate autoimmunity, H. pylori infection interferes with PC function by inducing alterations of mitochondrial biogenesis, which increases ROS damage and caspase-3-mediated apoptosis (green arrow), which leads to PC atrophy and inflammation
Our results show that the KI/KI2 mouse model and ATP4Ap.R703C patients do not overexpress IL-17. Therefore, PC atrophy did not occur (or at least, not exclusively) as a consequence of chronic exposure to the cytokine. Instead, the ATP4Ap.R703C mutation leads to a deregulation of the acid–base balance that triggers mitochondrial malfunction, which increases ROS damage signaling that fuels caspase-3-mediated apoptosis (Fig. 6b). Indeed, administering anti-IL-17A antibodies in the TxA23 mouse model (autoreactive to ATP4A-mediated atrophy) did not completely reverse parietal cell atrophy nor chronic gastritis [37], which is in agreement with our hypothesis. Alternatively, atrophy of PC might occur through an autophagy-like mechanism, where PC malfunction triggers CD4+ cells to activate caspase-3-mediated apoptosis. To prevent cellular damage, ROS-generating mitochondria are constantly removed by mitophagy, a specialized process of autophagy [28].
Inflammation and H. pylori infection
The ATP4Ap.R703C mutation impairs the internal acid–base balance in PCs, which deregulates mitochondrial DNA polymerase and the transcription factor Tfam. A recent study described that H. pylori infection also induces up-regulation of polg and Tfam to destabilize mtDNA cargo by saturation [23]. SPEM has also been observed in H. pylori patients [38].
Our results also show that H. pylori infection impairs acid–base balance and increases cell atrophy, cell cycle arrest and caspase-induced apoptosis (Fig. 4). The previous studies also demonstrated that H. pylori induces cell cycle arrest and apoptosis [39] and activation of the inflammasome [40]. Therefore, the mechanism by which H. pylori infection triggers atrophic gastric inflammation (including loss of parietal cells) may correlate with the genetic origin-mediated mechanism observed in the KI/KI2 mice and gNET patients (Fig. 6c). Adaptive immunity of certain epithelial cells (e.g., parietal cells) protects against infection by increasing gastric acid. Therefore, H. pylori infection may interfere with PC function to inactivate this protective mechanism.
Both H. pylori infection, which can progress to atrophy, pernicious anemia, metaplasia, and gastric cancer [41, 42], and our genetic-based autoimmune response activation mechanism may drive mitochondria deregulation, ROS activation, and, finally, cytokine production in the same manner (Fig. 6b, c). Importantly, mitochondrial function in the KI/KI2 mouse model was partially recovered when euchlorhydria was restored in prevention and reversion experiments (Fig. 2). This is also an important clue regarding new therapies for H. pylori eradication. The same response in H. pylori-infected patients would be expected after restoration of gastric acid levels to prevent atrophy of parietal cells and progression to gastritis. More detailed future studies are required to substantiate this important statement.
In summary, it is accepted that CD4+ T-mediated autoimmune chronic inflammation causes parietal cell atrophy and increases the risk of gastric neoplasms. However, our results demonstrate that the ATP4Ap.R703C mutation (genetic origin) found in patients affected with gastric neuroendocrine tumors (gNETs) leads to PC malfunction, which triggers an inflammatory response. The ATP4Ap.R703C mutation deregulates the acid–base balance inside parietal cells, which alters mitochondrial function and biogenesis. Mitochondrial malfunction activates ROS, which leads to oxidative stress-induced damage and caspase-3-mediated apoptosis and inflammation, which leads to atrophy of PCs. Therefore, genetic origin-mediated ATP4A malfunction initiates autoimmune disease; this contrasts with the classical description, according to which the autoimmune response causes PC atrophy. However, we cannot rule out other mechanisms of inflammation and autoimmunity affecting PC viability in other gastric pathologies different than genetic-mediated lesions. Mitochondrial malfunction was restored when KI/KI2 mice were treated with acidified water. Interestingly, H. pylori infection promotes achlorhydria by destabilizing PC mitochondria through a mechanism similar to the one described in APT4Ap.R703C patients, which suggests that restoration of gastric euchlorhydria might be indicated in these patients.
References
Schubert ML, Peura D. Control of gastric acid secretion in health and disease. Gastroenterology. 2008;134:1842–60.
Waldum HL, Sørdal ØF. The Enterochroma ffi n-like [ECL ] cell—central in gastric physiology and pathology. Int J Mol Sci. 2019;20:2444.
Schubert ML. Gastric physiology gastrointestinal anatomy and physiology. New York: Wiley; 2014. p. 58–77.
Coati I, Fassan M, Farinati F, Graham DY, Genta RM, Rugge M. Autoimmune gastritis: pathologist’s viewpoint. World J Gastroenterol. 2015;9:12179–89.
Nikou G, Angelopoulos T. Current concepts on gastric carcinoid tumors. Gastroenterol Res Pract. 2012;2012:287825.
Kidd M, Gustafsson BI. Management of gastric carcinoids (Neuroendocrine Neoplasms). Curr Gastroenterol Rep. 2012;14:467–72.
Calvete O, Reyes J, Zuñiga S, Paumard-Hernández B, Fernández V, Bujanda L, et al. Exome sequencing identifies ATP4A gene as responsible of an atypical familial type I gastric neuroendocrine tumour. Hum Mol Genetics. 2015;24:2914–22.
Calvete O, Herraiz M, Reyes J, Patiño A, Benitez J. A cumulative effect involving malfunction of the PTH1R and ATP4A genes explains a familial gastric neuroendocrine tumor with hypothyroidism and arthritis. Gastric Cancer. 2017;20:998–1003.
Calvete O, Varro A, Pritchard DM, Barroso A, Oteo M, Morcillo MÁ, et al. A knockin mouse model for human ATP4a R703C mutation identified in familial gastric neuroendocrine tumors recapitulates the premalignant condition of the human disease and suggests new therapeutic strategies. Dis Models Mech. 2016;9:025890.
Petersen CP, Mills JC, Goldenring JR. Murine models of gastric corpus preneoplasia. Levallois-Perret: CMGH; 2017. p. 11–26.
Correa P. A human model of gastric carcinogenesis 1. Cancer Res. 1988;48:3554–600.
Nguyen TL, Khurana SS, Bellone CJ, Capoccia BJ, Sagartz JE, Kesman RA Jr, et al. Autoimmune gastritis mediated by CD4+ T cells promotes the development of gastric cancer. Cancer Res. 2013;73:2117–266.
Bizzaro N, Antico A, Villalta D. Autoimmunity and gastric cancer. Int J Mol Sci. 2018;19:377.
Toh BH, Sentry JW, Alderuccio F. The causative H+/K+ATPase antigen in the pathogenesis of autoimmune gastritis. Immunol Today. 2000;21:348–54.
Hu Z, Chai J. Structural mechanisms in NLR inflammasome assembly and signalling. Current topics in microbiology and immunology. Cham: Springer; 2016. p. 23–42.
Chavarría-Smith J, Vance RE. The NLRP1 inflammasomes. Immunol Rev. 2015;265:22–34.
McHugh RS, Shevach EM, Margulies DH, Natarajan K. A T cell receptor transgenic model of severe, spontaneous organ-specific autoimmunity. Eur J Immunol. 2001;31:2094–103.
Bockerstett KA, Osaki LH, Petersen CP, Cai CW, Wong CF, Nguyen TLM, et al. Interleukin-17A promotes parietal cell atrophy by inducing apoptosis. Levallois-Perret: CMGH; 2018.
Amedei A, Bergman MP, Appelmelk BJ, Azzurri A, Benagiano M, Tamburini C, et al. Molecular mimicry between Helicobacter pylori antigens and H+, K+—adenosine triphosphatase in human gastric autoimmunity. J Exp Med. 2003;198:1147–56.
Shi Y, Liu XF, Zhuang Y, Zhang JY, Liu T, Yin Z, et al. Helicobacter pylori-Induced Th17 responses modulate Th1 cell responses, benefit bacterial growth, and contribute to pathology in mice. J Immunol. 2010;184:5121–9.
Hemminki K, Liu X, Ji J, Sundquist J, Sundquist K. Autoimmune disease and subsequent digestive tract cancer by histology. Ann Oncol. 2012;23:927–33.
Weis VG, Goldenring JR. Current understanding of SPEM and its standing in the preneoplastic process. Gastric Cancer. 2009;12:189–97.
Chatre L, Fernandes J, Michel V, Fiette L, Avé P, Arena G, et al. Helicobacter pylori targets mitochondrial import and components of mitochondrial DNA replication machinery through an alternative VacA-dependent and a VacA-independent mechanisms. Sci Rep. 2017;7:15901.
Judd LM, Andringa A, Rubio CA, Spicer Z, Shull GE, Miller ML. Gastric achlorhydria in H/K-ATPase-deficient (Atp4a(−/−)) mice causes severe hyperplasia, mucocystic metaplasia and upregulation of growth factors. J Gastroenterol Hepatol. 2005;20:1266–78.
Hayakawa Y, Fox JG, Wang TC. Isthmus stem cells are the origins of metaplasia in the gastric corpus. Levallois-Perret: CMGH; 2017. p. 89–94.
Burclaff J, Osaki LH, Liu D, Goldenring JR, Mills JC. Targeted apoptosis of parietal cells is insufficient to induce metaplasia in stomach. Gastroenterology. 2017;152:762–6.
He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41:1012–21.
Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–5.
Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411.
Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci. 2012;109:11282–7.
Bockerstett KA, DiPaolo RJ. Regulation of gastric carcinogenesis by inflammatory cytokines. Levallois-Perret: CMGH; 2017. p. 47–53.
Madej-Michniewicz A, Budkowska M, Salata D, Dolegowska B, Starzynska T, Blogowski W. Evaluation of selected interleukins in patients with different gastric neoplasms: a preliminary report. Sci Rep. 2015;5:14382.
Błogowski W, Madej-Michniewicz A, Marczuk N, Dołȩgowska B, Starzyńska T. Interleukins 17 and 23 in patients with gastric neoplasms. Sci Rep. 2016;6:37451.
Shen H-H, Yang Y-X, Meng X, Luo X-Y, Li X-M, Shuai Z-W, et al. NLRP3: A promising therapeutic target for autoimmune diseases. Autoimmun Rev. 2018;17:694–702.
Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Investig. 2007;117:60–9.
Howlett M, Chalinor HV, Buzzelli JN, Nguyen N, Van Driel IR, Bell KM, et al. IL-11 is a parietal cell cytokine that induces atrophic gastritis. Gut. 2012;61:1398–409.
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41.
Radyk MD, Burclaff J, Willet SG, Mills JC. Metaplastic cells in the stomach arise, independently of stem cells, via dedifferentiation or trans-differentiation of chief cells. Gastroenterology. 2018;154:839–43.
Canales J, Valenzuela M, Bravo J, Cerda-Opazo P, Jorquera C, Toledo H, et al. Helicobacter pylori Induced phosphatidylinositol-3-OH kinase/mTOR activation increases hypoxia inducible factor-1α to promote loss of cyclin D1 and G0/G1 cell cycle arrest in human gastric cells. Front Cell Infect Microbiol. 2017;7:92.
Latz E. The inflammasomes: mechanisms of activation and function. Curr Opin Immunol. 2010;22:28–33.
El-Zimaity H. Gastritis and gastric atrophy. Curr Opin Gastroenterol. 2008;24:682–6.
Fox JG, Wang TC, Rogers AB, Poutahidis T, Ge Z, Taylor N, et al. Host and microbial constituents influence Helicobacter pylori-induced cancer in a murine model of hypergastrinemia. Gastroenterology. 2003;124:1879–90.
Acknowledgements
We want acknowledge Fernando Escolar from CIB and Patricia González and Eduardo Caleiras from the Histopathology Unit Service at CNIO by their technical support. We also want to thank José Palacios and Cristian Perna from the Pathological Anatomy Service of the Ramón y Cajal Hospital for the transfer of samples. OC is granted by H2020 BRIDGES project (Number: 634935). JB’s lab is partially funded by CIBERER, by the Spanish Ministry of Health supported by FEDER funds (Grant Number PI16/00440), and by H2020 project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Human rights statement and informed consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964 and later versions. Informed consent to be included in the study, or the equivalent, was obtained from all patients.
Animal studies
All institutional and national guidelines for the care and use of laboratory animals were followed.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Benítez, J., Marra, R., Reyes, J. et al. A genetic origin for acid–base imbalance triggers the mitochondrial damage that explains the autoimmune response and drives to gastric neuroendocrine tumours. Gastric Cancer 23, 52–63 (2020). https://doi.org/10.1007/s10120-019-00982-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-019-00982-4