
Abstract
Sequence data was obtained from the capsid (ORF-2) and the polymerase (ORF-lb) genes of 23 turkey astrovirus (TAstV) isolates collected from commercial turkey flocks around the United States between 2003 and 2004. A high level of genetic variation was observed among the isolates, particularly in the capsid gene, where nucleotide sequence identity among them was as low as 69%. Isolates collected on the same farm, on the same day, but from different houses could have as little as 72% identity between their capsid gene sequences when compared. Phylogenetic analysis of the capsid gene revealed no clear assortment by geographic region or isolation date. The polymerase gene was more conserved with between 86 and 99% nucleotide identity and did assort in a geographic manner. Based on differing topologies of the capsid and polymerase gene phylogenetic trees, TAstV appears to undergo recombination.
Similar content being viewed by others

Astroviruses are small, non-enveloped, positive sense ssRNA viruses that are associated with enteric disease in humans [1] and numerous mammalian and avian species [1–4]. The astrovirus genome is approximately 7000 nt long and has a 5′ untranslated region followed by three open reading frames (ORF’s), a 3′ untranslated region, and a poly-A tail. Two ORF’s, ORF-1a and ORF-1b, are linked by a translational frame-shift and encode the non-structural proteins [5, 6]. Analysis of ORF-1b indicates that it encodes an RNA-dependent RNA polymerase [7]. This region of the astrovirus genome is the most conserved gene between the mammalian and the avian astroviruses, as well as among the avian astroviruses [8]. ORF-2 encodes the capsid protein, which is the primary antigenic determinant of the virus.
Turkey astrovirus (TAstV) was first described in 1980 by McNulty et al. [9] in the United Kingdom, and the first isolate of TAstV in the United States was identified in 1985 [10]. A second TAstV type, which was antigenically and genetically distinct from the previously identified United States isolates (TAstV-1), was isolated in 1996. This and similar isolates have been designated TAstV-2 [8, 11, 12]. The entire genome sequence of the prototype TAstV-2 isolate, NC/96, has been reported and bears many similar features to human astroviruses (HAstV’s) [11]. Based on epidemiological studies using electron microscopy as many as 78% of turkey flocks presenting enteritis have TAstV in their feces [13]. For this reason TAstV-2 is believed to be one of the etiological agents of poult enteritis complex, an economically important polymicrobial disease characterized by enteritis and, in severe cases, high mortality in commercial turkeys under 6 weeks of age [14].
In this study, the genetic diversity of TAstV-2 isolates collected in the US was evaluated by comparative analysis of the polymerase gene (ORF-1b) and capsid (ORF-2) gene sequences. Intestinal contents were collected from turkeys between 1 and 10 weeks of age from flocks in North Carolina (NC), Virginia (VA), California (CA) and Texas (TX) during 2003 and 2004. Specimens were collected primarily from flocks affected with enteritis, however, two isolates (NC/SEP/A40/03 and NC/SEP/A252/04) came from flocks described as healthy and performing well. Total RNA was extracted from intestinal contents by diluting 200 µl of the sample in 1.2 ml of PBS, homogenizing and performing a clarification by centrifugation. Then 250 µl of the supernatant was used for RNA extraction with Trizol LS reagent (Invitrogen Inc., Carlsbad, CA) in accordance with the manufacturer’s instructions.
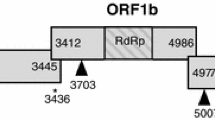
Samples were initially screened for TAstV by real-time reverse transcription-polymerase chain reaction (RRT-PCR) as previously described [15]. Portions of the polymerase (nucleotide position 4077–4880 by NC/96 numbering) and capsid genes (nucleotide position 6208–7035 by NC/96 numbering) of samples positive for TAstV by RRT-PCR were amplified by standard RT-PCR with the MKCap and MKPol primers as previously reported [16] (Fig. 1) with the Qiagen OneStep RT-PCR kit (Qiagen Inc., Valencia CA) in accordance with the kit instructions, without Q solution and with 0.6 µM of each primer. The region between the previously described segments of the polymerase and capsid genes of selected isolates was also amplified by RT-PCR. RT-PCR products were analyzed by agarose gel electrophoresis and amplicons of the correct size were excised, extracted with the QIAquick gel extraction kit (Qiagen Inc., Valencia CA) and directly sequenced with the BigDye terminator kit (Applied Biosystems, Foster City, CA) and were subsequently run on an ABI 3730 (Applied Biosystems, Foster City, CA). All RT-PCR products were also cloned with the TOPO TA cloning system (pCR2.1 vector) (Invitrogen Life Technologies, Chicago, IL) in accordance with the kit instructions. DNA plasmids containing the insert were prepared with the Qiaprep spin miniprep kit (Qiagen Inc., Valencia, CA). Plasmids were sequenced as described above.

Diagram of the turkey astrovirus genomic organization and location of primers used for amplification of segments of the polymerase and capsid genes. Numbers within the diagram represent the predicted start and stop sites. Primer sites are indicated with arrows
Sequences were aligned with ClustalW (DNASTAR Inc., Madison, WI) and phylogenetic analysis was performed with PAUP*4. 0b10 using the maximum parsimony tree building method with 1000 bootstrap replicates by heuristic search (Sinauer Associates Inc., Sunderland, MA). The following previously reported TAstV capsid and polymerase sequences from GenBank were included in the phylogenetic analysis: TAstV-1 complete genome (NC002470), TAstV-2 complete genome (NC005790) (NC/96), TAstV-2 polymerase partial sequence (AY320042) (TEVL-NC88), TAstV-2 capsid partial sequence (AY320042) (TEVL-NC88), and TAstV Ohio 2001 entire capsid sequence (AY769616).
Nucleotide (nt) and amino acid (aa) sequences of the polymerase gene were more conserved than the capsid gene among the TAstV isolates, which is consistent with what has been observed for HAstV’s [17, 18]. However, based on at least 10% nt divergence, the polymerase gene assorted into 2 groups (Fig. 2). Group 1 contained all recent isolates from Virginia (VA), North Carolina (NC) and Texas (TX), although the isolates from TX formed a sub-clade together. Among group 1 isolates there was nt similarity of between 94 and 100% and a predicted aa identity of between 97.5 and 100%. Group 2 consisted of all isolates from California (CA), and the two older NC isolates, NC/96 and TELV-NC88. This relationship suggests an epidemiological link, although no clear link is known. Within group 2 the nt similarity was between 96.7 and 98.9%. Between groups 1 and 2 there was at least 11.6% nt divergence. The nt identity with TAstV-1 was between 39.5 and 48.0% and the deduced aa identity between all isolates and TAstV-1 was less than 62.5%.

Phylogenetic tree of the (a) polymerase gene (ORF-1b) and (b) capsid gene (ORF-2) of TAstV’s isolated in the United States between 1988 and 2004. The tree was generated by the maximum likelihood method with heuristic search with PAUP*4.0b10 and is rooted with TAstV-1
Analysis of the nt and deduced aa sequences of the capsid region revealed substantial variation among the isolates (Fig. 2). Phylogenetically, the isolates assorted into 9 groups based on greater than 10% nt sequence divergence between groups. There was no clear assortment due to geographic origin or year of isolation. Within the groups the nt identity was as high as 99.7% (100% aa identity) and the most distantly related isolates, regardless of group, had 64.9% nt identity (73.6% aa identity). TAstV-1 had less than 47.8% aa identity with all the isolates studied.
Sequence variation among the capsid genes of TAstV was greater than expected and is higher than what has been reported for HAstV [19]. The sequence varies not only due to substitutions throughout the gene, but also with insertions and deletions which occur at several positions (Fig. 3). The Ohio 2001 isolate contained a unique 5 aa deletion. The isolates in phylogenetic groups 3, 4, 5 and 6 had a 2 aa insertion, and the 2 isolates from group 7 had a 4 aa insertion. This amount of variation would suggest that there is antigenic variation among the isolates, as Walter et al. [19] established that for HAstV that when two strains had less than 95% nucleotide homology they would constitute strains that could be serologically distinguished. However, due to the lack of an in vitro propagation system, antigenic analysis is unfeasible at this time.

ClustalW amino acid alignment of selected TAstV isolate capsid gene sequences. The positions where amino acids are identical are indicated as (.) and where amino acids are missing are indicated as (-). The boxed areas indicate amino acid insertions or deletions
Importantly, the topologies of the capsid and polymerase nt and aa trees differed. A particularly interesting example of this is given by the distinct assortment of the capsid and polymerase genes from isolates collected from different houses on the same farm, on the same day. Two sets of isolates fall into this category; the first being NC/SEP-A252/04 and NC/SEP-A253/04, and the second set being NC/SEP-A254/04 and NC/SEP-A255/04. The polymerase genes of each set assort together and share approximately 96% nt identity with the other isolate from the same farm. However, the capsid genes from the first set assort separately into groups 3 and 6 sharing only 85.1% nt identity, and the second set assort separately into groups 1 and 7 and have only 72.1% nt identity.
The irregular assortment of the polymerase and capsid genes of these and other individual viruses suggests that recombination may be occurring among TAstV isolates in the field. Genome recombination has been reported for several positive-sense, single-stranded RNA viruses including: astroviruses, coronaviruses, enteroviruses, and caliciviruses [17, 20–28]. To address the issue of recombination in more detail, ten isolates were sequenced in the intergenic region and analyzed. The 2.9 kb nucleotide sequence spanning the amplified region of the polymerase and capsid genes of four of these isolates (VA/SEP/A33/03, CA/SEP/A269/04, NC/SEP/A222/03 and TX/SEP/A311/04) and the TAstV-2 (NC/96) prototype was selected to demonstrate results. The SimPlot computer program [29] was used to analyze the alignment of the five TAstV isolates using a window size of 200 nucleotides that was moved along in 20-nucleotide steps. The percent identity was calculated for each window and plotted in a line chart. The chart showed that VA/SEP/A33/03 shared a low level of sequence identity in the polymerase region when compared to TAstV-2 NC/96 and a constantly high level of nt identity in the capsid region with the same virus. A single cross of the two curves was observed 200 nt after the ORF-1b/ORF-2 junction (Fig. 4). The likelihood analysis of recombination in DNA (LARD) program [30] was used to identify the point of recombination between VA/SEP/A33/03 and TAstV-2 NC/96. The predicted site was at nucleotide 1120 of the region amplified. This recombination site was different than the site suggested with other RNA viruses, where the recombination site is frequently found at the conserved region of the ORF’s junction [19, 24, 27]. When isolate CA/SEP/A269/04 and TAstV-2 NC/96 were compared, no significant change of sequence identity was observed between the polymerase and capsid regions. When isolates NC/SEP/A222/03 and TX/SEP/A311/04 were compared with TAstV-2 a low level of sequence identity in the polymerase and capsid regions was observed, although the junction region and the region from nucleotides 200 and 900 of the capsid gene had higher sequence identity.

Nucleotide identity plot of the 2.9 kb region comprising the amplified polymerase and capsid regions of TAstV-2 compared with TAstV isolates VA/SEP/A33/03 (33), NC/SEP/A222/03 (222), CA/SEP/A269/04 (269), TX/SEP/A311/04 (311). The bar below the plot represents the regions corresponding to ORF-1b (polymerase) and ORF-2 (capsid). The arrow indicates the point after the recombination site (nucleotide 1120) identified by LARD analysis
Recombination in ssRNA viruses is an important mechanism for generating diversity in enteroviruses such as polio [21], and also caliciviruses [27]. Walter et al. [20] reported that phylogenetic analysis of different regions of human astroviruses provided contradictory genotyping and demonstrated the occurrence of recombination between strains. Recombination requires concomitant infection of one host with two strains. Natural co-infection with two strains is certainly feasible due to the high incidence of TAstV-2 in commercial turkeys. The preferred mechanism of recombination among ssRNA viruses is a copy-choice mechanism where the polymerase enzyme switches from copying one RNA molecule (donor template) to another (acceptor template) without releasing the nascent strand [31].
Antigenic variation among astroviruses in different species has been well described. Eight serotypes of HAstV and three serotypes of bovine astrovirus have been defined [1] and are believed to evolve to some extent as immunological escape mutants. Interestingly, turkeys do not seem to elicit a good antibody response to the TAstV [32], which may explain why the virus circulates as multiple discrete sub-lineages similar to HIV [33, 34] as opposed to homogenous populations which undergo gradual antigenic drift with host immune pressure thereby assorting chronologically and geographically [35, 36].
In conclusion, comparative analysis of 23 TAstV isolate capsid genes revealed nine distinct genotypes circulating in the US during 2003 and 2004. Multiple TAstV genotypes were often present in one geographical region and even on the same farm. Although the polymerase gene assorted into two phylogenetic groups, this gene was more conserved and may be able to serve as a target for nucleic acid detection tests. Furthermore, evidence of recombination was shown by different topologies of the polymerase and capsid gene phylogenetic trees and by SimPlot analysis. It is also interesting to note that the two isolates collected from healthy flocks grouped together. Finally, extensive genetic variation, and the likely consequential antigenic variation, has practical implications for virus detection methods, vaccine development and epidemiological studies.
References
S.M. Matsui, H.B. Greenberg, in Fields Virology, 4th edn, ed. by D.M. Knipe, P.M. Howley (Lippincott Williams and Wilkins, Baltimore, MD, 2001)
R.E. Gough, M.S. Collins, E. Borland, L.F. Keymer, Vet. Rec. 114(11), 279 (1984)
T. Imada, S. Yamaguchi, M. Mase, K. Tsukamoto, M. Kubo, A. Morooka, J. Virol. 74(18), 8487–8493 (2000)
W. Baxendale, T. Mebatsion, Avian Pathol. 33(3), 364–370 (2004)
C.A. Gibson, J. Chen, S.A. Monroe, M.R. Denison, Adv. Exp. Med. Biol. 440, 387–391 (1998)
B. Jiang, S.S. Monroe, E.V. Koonin, S.E. Stine, R.I. Glass, Proc. Natl. Acad. Sci. USA 90(22), 10539–10543 (1993)
S.M. Matsui, D. Kiang, N. Ginzton, T. Chew, U. Geigenmuller-Gnirke, Novartis Found. Symp. 238, 219–233 (2001) discussion 33–36
M.D. Koci, S. Schultz-Cherry, Avian Pathol. 31(3), 213–227 (2002)
M.S. McNulty, W.L. Curran, J.B. McFerran, Vet. Rec. 106(26), 561 (1980)
L.J. Saif, Y.M. Saif, K.W. Theil, Avian Dis. 29(3), 798–811 (1985)
M.D. Koci, B.S. Seal, S. Schultz-Cherry, J. Virol. 74(13), 6173–6177 (2000)
S. Schultz-Cherry, D.J. King, M.D. Koci, Avian Dis. 45(1), 76–82 (2001)
D.L. Reynolds, Y.M. Saif, K.W. Theil, Avian Dis. 31(1), 89–98 (1987)
H.J. Barnes, J.S. Guy, J.P. Vaillancourt, Rev. Sci. Tech. 19, 565–588 (2000)
E. Spackman, D. Kapczynski, H. Sellers, Avian Dis. 49(1), 86–91 (2005)
M.D. Koci, B.S. Seal, S. Schultz-Cherry, J. Virol. Methods 90(1), 79–83 (2000)
G. Belliot, H. Laveran, S.S. Monroe, Arch. Virol. 142(7), 1323–1334 (1997)
T.L. Lewis, H.B. Greenberg, J.E. Herrmann, L.S. Smith, S.M. Matsui, J. Virol. 68(1), 77–83 (1994)
J.E. Walter, D.K. Mitchell, M.L. Guerrero, T. Berke, D.O. Matson, S.S. Monroe, et al J. Infect. Dis. 183(5), 681–686 (2001)
J.E. Walter, J. Briggs, M.L. Guerrero, D.O. Matson, L.K. Pickering, G. Ruiz-Palacios, et al Arch. Virol. 146(12), 2357–2367 (2001)
O. Kew, V. Morris-Glasgow, M. Landaverde, C. Burns, J. Shaw, Z. Garib, Science 296(5566), 356–359 (2002)
M.S. Oberste, K. Maher, M.A. Pallansch, J. Virol. 78(2), 855–867 (2004)
M.S. Oberste, S. Penaranda, M.A. Pallansch, J. Virol. 78(6), 2948–2955 (2004)
S.L. Oliver, D.W. Brown, J. Green, J.C. Bridger, Virology 326(2), 231–239 (2004)
S.L. Oliver, A.M. Dastjerdi, S. Wong, L. El-Attar, C. Gallimore, D.W. Brown, et al J. Virol. 77(4), 2789–2798 (2003)
G. Oprisan, M. Combiescu, S. Guillot, V. Caro, A. Combiescu, F. Delpeyroux, et al. J. Gen. Virol. 83(Pt 9), 2193–2200 (2002)
X. Jiang, C. Espul, W.M. Zhong, H. Cuello, D.O. Matson, Arch. Virol. 144(12), 2377–2387 (1999)
L.R. Banner, M.M.-C. Lai, Virology 185, 441–445 (1991)
K.S. Lole, R.C. Bollinger, R.S. Paranjape, D. Gadkari, S.S. Kulkarni, N.G. Novak, et al J. Virol. 73(1), 152–160 (1999)
E.C. Holmes, M. Worobey, A. Rambaut, Mol. Biol. Evol. 16, 405–409 (1999)
M.M.-C. Lai, Microbiol. Rev. 56(1), 61–79 (1992)
M.D. Koci, L.A. Kelley, D. Larsen, S. Schultz-Cherry, J. Virol. 78(3), 1564–1574 (2004)
A. Rambaut, D. Posada, K.A. Crandall, E.C. Holmes, Nat. Rev. Genet. 5(1), 52–61 (2004)
M.A. Papathanasopoulos, G.M. Hunt, C.T. Tiemessen, Virus Genes 26(2), 151–163 (2003)
J.B. Plotkin, J. Dushoff, S.A. Levin, Proc. Natl. Acad. Sci. USA 99(9), 6263–6268 (2002)
D.J. Smith, A.S. Lapedes, de J.C. Jong, T.M. Bestebroer, G.F. Rimmelzwaan, A.D. Osterhaus, et al Science 305(5682), 371–376 (2004)
Acknowlegements
We gratefully acknowledge our industry partners for providing the specimens that made this study possible. We also thank Diane Smith, Scott Lee, Dawn Williams-Coplin and the SAA sequencing facility for technical assistance with this work. The work was supported by USDA/ARS CRIS project #6612-32000-036.
Author information
Authors and Affiliations
Corresponding author
Additional information
Nucleotide sequence accession numbers. The TAstV nucleotide sequences presented in this article have been submitted to GenBank under Accession Numbers: DQ066553–DQ066575 for the capsid sequences and DQ066576–DQ066598 for the polymerase sequences. Mention of trade names or commercial products in this manuscript is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture.
Rights and permissions
About this article
Cite this article
Pantin-Jackwood, M.J., Spackman, E. & Woolcock, P.R. Phylogenetic Analysis of Turkey Astroviruses Reveals Evidence of Recombination. Virus Genes 32, 187–192 (2006). https://doi.org/10.1007/s11262-005-6875-3
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s11262-005-6875-3