
Abstract
Multiple myeloma (MM) is an incurable malignancy of plasma cells with a clinical course characterized by multiple relapses and treatment refractoriness. While recent treatment advancements have extended overall survival (OS), refractory MM has a poor prognosis, with a median OS of between 4 and 6 months. Nuclear export inhibition, specifically inhibition of CRM1/XPO1, is an emerging novel treatment modality that has shown promise in treatment-refractory MM. Initially discovered in yeast in 1983, early clinical applications were met with significant toxicities that limited their utility. The creation of small molecule inhibitors of nuclear export (SINE) has improved on toxicity limitations and has led to investigation in a number of malignancies at the preclinical and clinical stages. Preclinical studies of SINEs in MM have shown that these molecules are cytotoxic to myeloma cells, play a role in therapy resensitization, and suggest a role in limiting bone disease progression. In July 2019, selinexor became the first nuclear export inhibitor approved for use in relapsed/refractory MM based on the STORM trial. As of May 2020, there were eight ongoing trials combining selinexor with standard treatment regimens in relapsed/refractory MM. Eltanexor, a second-generation SINE, is also under investigation and has shown preliminary signs of efficacy in an early clinical trial while potentially having an improved toxicity profile compared with selinexor. Results in ongoing trials will help further define the role of SINEs in MM.
Similar content being viewed by others

Multiple myeloma (MM) cells have been shown to have increased nuclear export protein expression that has been associated with increased lytic lesions as well as shorter progression-free survival and overall survival. |
Selinexor, an inhibitor of nuclear export, has shown benefit in treatment-refractory MM when used alone or in combination with dexamethasone or other conventional MM therapies. |
Results from numerous clinical trials evaluating selinexor in MM are eagerly anticipated to help define the role of nuclear export inhibition in MM. |
1 Introduction
Multiple myeloma (MM) is a plasma cell malignancy treated with combinations of drugs from a variety of drug classes, including immunomodulatory drugs (IMiDs; thalidomide, lenalidomide, pomalidomide), proteasome inhibitors (PIs; bortezomib, carflizomib, ixazomib), anti-CD38 monoclonal antibodies (daratumumab, isatuximab), pan-deacetylase inhibitor (panobinostat), or immunostimulatory anti-SLAMF7 antibody (elotuzumab), in addition to cytotoxic chemotherapy, corticosteroids, and autologous stem cell transplantation [1, 2]. Despite this plethora of treatment modalities, relapses are inevitable and remission durations become progressively shorter. When patients eventually become triple-class refractory (i.e. refractory to IMiDs, PIs, and anti-CD38 monoclonal antibodies), median overall survival (OS) can be as short as 4–6 months [3]. Novel approaches are urgently needed for patients who are refractory to available treatments.
Targeting nuclear export for anticancer therapy is an emerging field that has shown promise in MM. Regulation of the cell cycle is in part mediated by proteins facilitating the transport of molecules across the nuclear envelope. Chromosomal maintenance 1 (CRM1), also known as exportin 1 (XPO1), is a protein that interacts with a nuclear export signal and mediates the export of proteins and RNA from the nucleus into the cytoplasm [4]. Aberrant regulation of this nuclear-cytoplasmic transport has been implicated in the development of cancer. Overexpression of XPO1 results in the mislocation of tumor suppressor proteins such as p53, APC/β-catenin, FOXO3, BRCA 1/2, IkBa, survivin, and others. In some cancers, DNA topoisomerases I and IIa are shuttled into the cytoplasm via XPO1. This ultimately prevents anthracycline- and etoposide-induced cell death, which requires the topoisomerases to remain intranuclear. Oncogenes such as BCR-ABL, and oncoproteins such as c-myc, are also shuttled via XPO1, which acts to increase their oncogenic potential [5, 6]. In addition, high XPO1 expression has been observed to be a poor prognostic indicator in some cancers such as acute myeloid lymphoma (AML) and gastric cancer [7, 8]. A number of nuclear export inhibitors are now under investigation in a range of malignancies (Table 1).
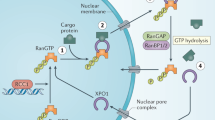
Knockdown studies of XPO1 in myeloma cells have revealed its importance to myeloma cell survival, hinting at its potential as a therapeutic target [9]. Gene expression analyses have shown increased XPO1 expression in MM cells compared with plasma cells from healthy subjects and those with monoclonal gammopathy of undetermined significance (MGUS) and smoldering MM (SMM) [10]. Similarly, expression is higher in human myeloma cell lines than primary samples from MM patients. Increased XPO1 expression is associated with increased lytic lesions, as well as shorter progression-free survival (PFS) and OS [11]. XPO1 expression also plays a role in drug resistance, with XPO1 having a fourfold increased expression in bortezomib-resistant MM cell lines [12]. Of note, corticosteroids have well known anti-myeloma activity, and transport of the glucocorticoid receptor (GR) is also mediated by XPO1. Research into XPO1 inhibition has led to the development of selinexor, the first US FDA-approved inhibitor of nuclear export that is now approved in penta-refractory MM (i.e. patients refractory to at least two immunomodulatory agents, two proteasome inhibitors, and an anti-CD38 monoclonal antibody) (Fig. 1) [13].

Nuclear export inhibition with selinexor (used with permission from Karyopharm Therapeutics). a Nuclear export before inhibition with selinexor. Uninhibited nuclear export results in the shuttling of oncoprotein messenger RNA (mRNA) and tumor suppressor proteins from the nucleus into the cytoplasm. b Inhibition of nuclear export with selinexor. Inhibition of nuclear export with selinexor results in the trapping of tumor suppressor proteins and oncoprotein mRNA into the cell nucleus
2 Development of Natural Nuclear Export Inhibitors and Small Inhibitors of Nuclear Export
2.1 Leptomycin B and Ratjadone C
The first discovered inhibitor of nuclear export via XPO1 inhibition was leptomycin B (LMB). Originally isolated from Streptomyces sp. ATS1287, LMB was discovered during a program searching for new antifungal antibiotics in 1983 [14]. The relation of LMB to XPO1 was first established in 1993 by Nishi et al. during their study of LMB resistant Schizosaccharomyces pombe [15]. They showed that mutations in XPO1 caused LMB resistance, and amplification of wild-type XPO1 conferred LMB resistance in wild-type S. pombe. In 1999, Kudo et al. discovered that LMB covalently binds and alkylates a cysteine residue on XPO1, leading to XPO1 inactivation via blocking of the nuclear export sequence of cargo proteins [16]. LMB was found to have significant activity against a variety of experimental leukemia, melanoma, sarcoma, and other tumor models [17]. In 1996, a phase I clinical trial of LMB (termed Elastocin) was initiated at the Charing Cross Hospital in London [18]. A total of 33 patients with diagnoses including colon, ovary, melanoma, glioma, sarcoma, pancreas, and other cancers were enrolled. Significant toxicities were observed, including nausea, vomiting, severe anorexia, and malaise, with ultimately no partial response seen in these patients.
Ratjadone C, isolated from myxobacterium Sorangium cellulosum, is another XPO1 inhibitor of similar structure and molecular mechanism to LMB that has been tested in vitro only [4]. In 2003, Burzlaff et al. found ratjadone inhibited growth against the tumor lines Jurkat, HepG2, and U87-MG [19]. Using MM cell lines, Turner et al. demonstrated that Ratjadone C was able to sensitize these cells to doxorubicin and etoposide by blocking topoisomerase II from being shuttled out of the nucleus via XPO1 [20].
2.2 Curcumin
Curcumin is a polyphenol compound found in turmeric and has been used in traditional medicines for thousands of years for its proposed anti-inflammatory and anticancer properties [21]. While numerous mechanisms of action have been proposed, Mingshan et al. demonstrated that curcumin targets XPO1 and inhibits nuclear export [22]. Unlike other nuclear export inhibitors, curcumin also inhibits the nuclear export of p53 by blocking the phosphorylation of p53 by Jun activation-domain binding protein (Jab1) [23]. Curcumin has been tested against numerous human myeloma cell lines with success [24]. Cells lines with poor prognostic indicators t(4;14) and t(14;16) were found to be sensitive to curcumin, with sensitivity being independent of TP53 status. Primary myeloma cells, including those with del(17p), were also found to be sensitive to curcumin. In patients with MGUS and SMM, investigators have found curcumin therapy resulted in decreased bone turnover and paraprotein load, suggesting curcumin may slow disease progression [25]. In 2020, Ramakrishna et al. used curcumin in place of dexamethasone in 15 MM patients age > 55 years who were no longer tolerating dexamethasone [26]. Curcumin C3 complex, at 3–4 g daily, was administered alongside either an IMiD or PI, resulting in a reduction in paraprotein load by 38% and plasmacytosis by 59%. The major adverse effect was diarrhea, which improved with cessation or by decreasing the dose of curcumin. In an effort to improve on the poor oral bioavailability of curcumin, intravenous formulations of curcumin have been developed. In 2019, SignPath Pharma demonstrated that their formulation of liposomal curcumin (LipoCurc) had intense uptake in MM cell lines compared with red blood cells and mononuclear cells, with minimal metabolism to tetrahydrocurcumin (THC), the main inactive metabolite [27]. The maximum tolerated dose has been established and phase I/II trials in MM are currently under development [28]. Ultimately, more studies are needed to further characterize the mechanism of action of curcumin, specifically as it relates to MM, its clinical efficacy, and how it may fit in the myeloma treatment paradigm.
2.3 SL-801 (CBS9106, Felezonexor)
The toxicity found in the LMB phase I trial led to the study of a synthetic XPO1 inhibitor, CBS9106, by Sakakibara et al. [29]. In vitro, CBS9106 was tested against over 60 human cell lines, including MM lines MM.1S and RPMI-8226. CBS9106 was found to suppress cell growth when used alone and sensitized these cells to radiation. CBS9106 and LMB both led to the inhibition of tumor necrosis factor (TNF)-α-induced IκB-α degradation in MM.1S and RPMI-8226 cells, and it was postulated that this could be a mechanism by which CBS9106 inhibits MM cell growth. In mouse xenograft models, CBS9106 was well tolerated, with no significant weight loss, and showed statistically significant antitumor activity and prolongation of survival time.
A phase I clinical trial using CBS9106 (termed SL-801) in patients with advanced solid tumors was initiated in 2016 (NCT02667873). The study aims to enroll 70 participants with metastatic or locally advanced and unresectable solid tumors that are resistant to standard therapy or if non-standard and radiation therapies are not treatment options. Interim results released in October 2019 revealed a partial response in a patient with KRAS+ microsatellite stable colorectal cancer after two cycles [30]. Stable disease was achieved in 12 patients, with 20% disease shrinkage seen in a patient with a heavily pretreated neuroendocrine tumor. Of the released data, treatment-related adverse events included nausea, vomiting, fatigue, decreased appetite, diarrhea, acute renal injury, and neutropenia.
3 Small Molecule Inhibitors of Nuclear Export (SINE)
The most promising of the XPO1 inhibitors is a class known as small molecule inhibitors of nuclear export (SINE). Karyopharm Therapeutics identified numerous diverse novel SINE compounds using a method of structure-based drug design termed consensus-induced fit docking (cFID) [31]. Of these SINEs, KPT-330, otherwise known as selinexor, has demonstrated the most success due to its superior bioavailability and potency [32]. In preclinical studies, selinexor reduced proliferation and induced growth inhibition and/or apoptosis in MM, AML, chronic lymphocytic lymphoma (CLL), lymphoma, renal, prostate, breast, ovarian, colorectal liver, pancreatic, non-small cell lung cancer, thyroid, sarcoma, mesothelioma, glioma, and melanoma malignancies [33]. Recently, based on data that XPO inhibition also blocks viral replication and ensuing inflammation, a randomized phase II clinical trial of low-dose selinexor versus placebo in patients hospitalized with severe coronavirus disease 2019 (COVID-19) was also initiated (NCT04349098).
3.1 Selinexor
The use of selinexor in preclinical studies of MM has demonstrated its ability to work synergistically with chemotherapeutic agents and dexamethasone. In 2013, Turner et al. demonstrated that SINEs, including selinexor, induced apoptosis both as a single agent or in a synergistic manner when combined with doxorubicin, bortezomib, or carfilzomib in myeloma cell populations [34]. This effect was observed to be dose-dependent and spared normal peripheral blood mononuclear cells. Co-incubation with doxorubicin was observed to induce activated caspase 3 in myeloma, but not non-myeloma, cell populations. These findings suggested that SINEs may be selective specifically for myeloma cells, unlike LMB in prior studies. The sensitization of myeloma cells to doxorubicin, bortezomib, and carfilzomib in the presence of selinexor was further supported in later studies [35,36,37,38]. In 2014, Tai et al demonstrated that not only do SINEs induce apoptosis but they also block receptor activator of nuclear factor kappa-Β ligand (RANKL)-induced nuclear factor (NF)-kB and NFATc1, key osteoclast differentiation regulators [39]. The blockage of NFATc1 and downstream differentiation genes prevented adhesion and formation of functional osteoclasts, indicating that SINEs also have the added benefit of reducing the progression of bone disease. In 2018, Argueta et al. demonstrated that selinexor has synergistic effects with dexamethasone in a GR-dependent manner [40]. Selinexor enhances the transcription and translation of the GR while dexamethasone activates GR, which ultimately leads to antitumor activity and cell death. It was suggested that selinexor can be used to resensitize patients to dexamethasone or be beneficial in patients who lack GR activity. Mammalian target of rapamycin (mTOR), a molecule that contributes to the progression of myeloma, was inhibited with this combination, in part through enhancing the expression of the negative regular REDD1. In a xenograft model of severe combined immunodeficient (SCID) mice, the authors found that selinexor–dexamethasone-treated mice had significantly reduced tumor growth compared with mice treated with selinexor or dexamethasone alone. A number of clinical trials are currently underway for the use of selinexor in multiple malignancies.
3.1.1 Selinexor in Multiple Myeloma (MM)
In a phase I dose-escalation study, selinexor 3–60 mg/m2 without dexamethasone had limited activity in patients with MM and Waldenstrom macroglobulinemia [41]. A dose of 80 mg + dexamethasone 20 mg twice weekly was associated with a 50% overall response rate (ORR; n = 12, not daratumumab-exposed or quad-refractory). The half-life was 6–8 h, although the pharmacologic half-life based on XPO messenger RNA (mRNA) expression was 48 h. The recommended phase II dose (RP2D) was determined to be selinexor 80 mg plus dexamethasone 20 mg administered twice weekly.
On 3 July 2019, the FDA granted accelerated approval for selinexor, the first time an XPO1 inhibitor has been approved for use in MM. Approval was ultimately based on the STORM study, a phase IIb, multicenter, open-label study taking place between May 2015 and March 2018 in the US and Europe [41]. Selection criteria included previous treatment with bortezomib, carfilzomib, lenalidomide, pomalidomide, daratumumab, glucocorticoids, and an alkylating agent; disease refractoriness to one or more immunomodulatory agent, PI, daratumumab, glucocorticoids, and their most recent regimen; creatinine clearance ≥ 20 mL/min, absolute neutrophil count ≥ 1000/mm, platelets ≥ 75,000/mm3 (if bone marrow plasma cell > 50%; platelets > 50,000/mm3), and hemoglobin ≥ 8.5 g/dL. The primary endpoint was overall response, with secondary endpoints being response duration, PFS, and OS. A total of 122 patients with progressive myeloma met the eligibility criteria, with 117 (96%) patients being refractory to all three of the most potent drugs in each class (i.e. pomalidomide, carfilzomib, and daratumumab). Refractoriness to therapy was defined as a response of stable disease or worse, or relapse within 60 days of discontinuing treatment. The median age was 65.2 years, with 53% of patients having high-risk cytogenetic abnormalities, with a median of seven previous therapies during the 6.6 years since diagnosis, indicating functionally high-risk disease. There was also a median 22% increase in the monoclonal protein during a median of 12 days from the day of consent to cycle 1, day 1, demonstrating the rapidly progressive nature of triple-class refractory MM.
Selinexor (80 mg) along with dexamethasone (20 mg) was administered weekly on days 1 and 3 within 4-week cycles until disease progression, discontinuation, or death [41]. All patients received ondansetron 8 mg before the first dose, with other supportive measures administered as needed. The median time to response was 4.1 weeks, including two patients who had progressed after prior chimeric antigen receptor T-cell therapy (CAR-T). Overall response, defined as partial response or better, was 26% (n = 32); 13% achieved minimal response, 39% had stable disease, and 21% had progressive disease. Median PFS was 3.7 months and OS was 8.6 months. Of the patients enrolled, 96% discontinued treatment, usually due to adverse events (19.5%) or disease progression (55.1%). The most common adverse events were thrombocytopenia (73%), fatigue (73%), and nausea (72%). Common grade 3/4 adverse events included thrombocytopenia (59%), anemia (44%), and hyponatremia (22%) (Table 2). In the center with the highest enrollment (n = 28), despite comparable baseline characteristics and overall rates of AEs, the ORR, PFS, and OS were 53.6%, 5.3 months, and 15.6 months, respectively, likely due to only two patients coming off for toxicity (manuscript in preparation). At this center, an aggressive, multiagent, antinausea prophylaxis (ondansetron, NK1 receptor antagonist, and olanzapine), close symptom and laboratory monitoring during cycle 1 with supportive care, and dose holds/modification were used. Therefore, in a population with rapidly progressive disease, higher doses of selinexor are needed initially to attain disease control (lower doses were not as efficacious in the phase I dose-escalation study) with aggressive supportive care, and, thereafter, the selinexor dose typically ends up being lowered to a maintenance dose. Of note, putting aside the selinexor-associated increase in GR activity, it is especially important to be aware that the antiemetic aprepitant, an NK1 receptor antagonist (NK1RA), is a moderate inhibitor of cytochrome P450 isoenzyme 3A4. As a result, there is a 2.2-fold increase in the area under the concentration–time curve of dexamethasone, therefore either the dose of dexamethasone must be reduced by 50% or, alternatively, the NK1RA rolapitant, which does not have this interaction, can be used [42].
Recent data from the registrational phase III BOSTON trial evaluating selinexor 100 mg in combination with once-weekly bortezomib and dexamethasone (SVd) to a control arm of twice-weekly bortezomib and dexamethasone (Vd) were presented at ASCO 2020 [43]. In 402 patients with relapsed/refractory MM (RRMM) who previously received between one and three prior treatment regimens (NCT03110562), in spite of 40% lower bortezomib and 25% lower dexamethasone doses at 24 weeks (eight cycles), the study met its primary endpoint, with a median PFS of 13.93 months and 9.46 months (hazard ratio 0.70; p = 0.0066) for SVd and Vd, respectively. Interestingly, approximately 50% of patients in both arms were high risk [central fluorescent in situ hybridization (FISH) testing and del (17p) or t(14;16) or t(4;14) or amp 1q21] and the PFS hazard ratio for high risk (0.67 [0.45‒0.98]) was comparable with the standard risk 0.62 (0.42‒0.95). For the 37 patients with del 17p, the hazard ratio was even lower at 0.38 (0.16‒0.86). No imbalance in deaths between the two groups was reported and the median OS was not reached in both arms, although OS will be difficult to interpret as crossover was permitted in this study for those patients progressing on Vd to receive SVd; however, more patients came off for either adverse events or patient withdrawal (36% and 20% in SVd and Vd, respectively) [45]. There were higher rates of cytopenia, gastrointestinal issues, weight decrease, fatigue, and cataract in the SVd arm, whereas neuropathy rates were higher in the Vd arm. The increased frequency of cataracts in the SVd arm also raises the possibility of increased corticosteroid activity (despite a lower dose of dexamethasone in the SVd arm). While cross-study comparisons are fraught with issues, especially here where the STORM trial included a much more heavily treated population treated with selinexor 80 mg twice weekly, whereas the BOSTON trial included a less heavily treated population treated with 100 mg weekly, the AEs are generally lower in the BOSTON trial, especially considering the contribution of bortezomib in the BOSTON trial.
As demonstrated by the BOSTON study, given the genomic complexity of RRMM, combination therapy is typically more efficacious than single-agent or doublet regimens. That said, given the tolerability issues with bortezomib, there is greater interest in other combination strategies. The STOMP study has demonstrated that the recommended dosing of selinexor with bortezomib, carfilzomib, or daratumumab is 100 mg weekly, whereas with lenalidomide or pomalidomide, dosing is 60 mg weekly due to overlapping hematologic toxicities. The response rates with these triplets can range from approximately 50–80% and toxicities are those seen with the individual agents. Of note, the responses in the post CAR-T setting have been validated in other patients who had responses with selinexor containing triplet regimens, with remissions lasting nearly as long as their CAR-T remissions [44].
3.1.2 Ongoing Selinexor Trials in MM
There are currently over 40 ongoing trials using selinexor in numerous different malignancies, and eight active trials of selinexor in MM listed on the National Institutes of Health clinical trials database (Table 3).
3.2 Eltanexor
Eltanexor, also known as KPT8602, is a next-generation XPO1 inhibitor that has shown some promise in reducing toxicity compared with selinexor. Eltanexor binds to XPO1 and inhibits XPO1–cargo interactions in a similar manner to previous generation SINEs. Hing and colleagues demonstrated that eltanexor and selinexor have similar cytotoxicity in CLL, AML, and diffuse large B-cell lymphoma (DLBCL) representative cell lines [51]. Eltanexor was better tolerated and was shown to have reduced CNS penetration in mouse, rat, and monkey models. In CLL and patient-derived xenograft AML mouse models, eltanexor was demonstrated to prolong survival. Using numerous MM cell lines, Turney et al. demonstrated that eltanexor led to apoptosis and functioned synergistically in all cell lines when combined with common anti-MM agents such as bortezomib, carfilzomib, doxorubicin, melphalan, and etoposide [52]. Using mouse models, the authors did not observe weight loss toxicity that is typically observed with selinexor. Treated MM cells from newly diagnosed/relapsed patient bone marrow aspirates with eltanexor + typical anti-MM agents revealed that combination treatment was more effective than a single agent in inducing apoptosis.
Currently, a phase I/II clinical trial sponsored by Karyopharm Therapeutics, Inc. is underway to evaluate the safety, tolerability, and efficacy of eltanexor in relapsed/refractory MM (RRMM), metastatic colorectal cancer, metastatic castration-resistant prostate cancer, and higher risk myelodysplastic syndrome (NCT02649790). In 2017, preliminary data of the MM arm were released. Inclusion criteria in this group included patients with confirmed symptomatic RRMM that was previously treated and refractory to three or more therapies, including an IMiD, PI, alkylator, and corticosteroid [53]. This arm was designed as a 3 + 3 dose escalation study. Oral eltanexor was administered as 5–60 mg (± dexamethasone) either every other day for 3 days per week or daily during a 28-day cycle. A total of 34 evaluable patients were reported, with a median time on treatment of 96 days. The best responses were observed in patients receiving 20 and 30 mg plus dexamethasone, with an ORR of 35%, clinical benefit rate (CBR) of 64%, and progressive disease rate (PR) of 7.1%. This was compared with an ORR of 21%, CBR of 47%, and PR of 18% among all patients. Nausea (54%), fatigue (46%), anemia (38%), diarrhea (38%), weight loss (33%), and neutropenia (31%) were the most common grade 1/2 adverse effects. Grade 3/4 adverse effects included thrombocytopenia (56%), neutropenia (26%), anemia and leukopenia (15%), and hyponatremia (10%). More patients had decreased appetite and weight loss at and above 30 mg. Dose escalation was halted as efficacy was reached, therefore maximum tolerated dose was not determined. Based on the greater efficacy and improved adverse effect profile, the RP2D of eltanexor was established to be 20 mg administered five times per week with 20 mg of dexamethasone administered twice weekly. Overall, these preliminary data have shown eltanexor can be efficacious in MM while potentially having a superior adverse effect profile to selinexor.
4 Discussion and Conclusions
The initial treatment of MM is guided by host factors (age, comorbidities, and functional status), disease factors (symptom severity, International Staging System [ISS] stage, and molecular risk), and treatment factors (efficacy/toxicity, route of administration, and availability/cost). General standards of care have been established in initial therapy, with various combinations of lenalidomide, bortezomib, daratumumab, and dexamethasone used in most initial regimens regardless of transplant eligibility [54]. Ultimately, nearly all patients relapse, with ever-decreasing remission durations with each relapse. The choice of treatment at relapse is determined by all considerations at the initial diagnosis and also the time of relapse and tolerance of and response/refractoriness to prior therapy. Today, combinations of IMiDs, PIs, anti-CD38 antibodies, pan-deacetylase inhibitors (panobinostat) and anti-SLAMF7 antibodies (elotuzumab) are available at relapse. Despite numerous options, patients typically become refractory to all drug classes [55]. The treatment of this multidrug refractory population is an unmet need. Data from approximately 7400 patients treated with 129 drugs in 228 early-phase studies demonstrated that the threshold response rate needed for regulatory approval and widespread clinical use is 20%, however this gets increasingly challenging for a single agent (± dexamethasone) to achieve in increasingly drug-refractory patients.
Nuclear export inhibition is a novel therapeutic mechanism in MM, with strong preclinical rationale based on XPO1 overexpression in MM cells and correlation with worse clinical outcomes. Preclinical trials in nuclear export inhibition have shown direct cytotoxicity in myeloma cell lines and suggest possible therapy resensitization. Clinical trials have shown this new drug class to be efficacious when used alone or in combination with dexamethasone or other conventional agents used in MM. In MM patients who experience multiple relapses, selinexor provides a new treatment option in those refractory to at least two immunomodulatory agents, two PIs, and an anti-CD38 monoclonal antibody. In this population, with rapidly progressive disease and rather permissive eligibility criteria (e.g. creatinine clearance of 20 mL/min and an ANC of 1000/uL), the STORM trial demonstrated an ORR of 26%, a median PFS of 3.7 months, and a median OS of 8.6 months. These results distinguish selinexor from panobinostat, elotuzumab, and ixazomib, which required combination regimens in studies for approval, and put it more in the category of carfilzomib, pomalidomide, and daratumumab, which all also received accelerated approval due to activity demonstrated in a single-arm study in a population with no other available treatment options.
The STORM study also demonstrated significant grade 3/4 toxicity, primarily in four categories—fatigue, gastrointestinal (particularly nausea/vomiting, decreased appetite, weight loss), hyponatremia, and hematologic toxicities. However, toxicities are also in part due to the patient population. For example, the incidence of thrombocytopenia is highest in patients with triple-class refractory myeloma (58% with grade 3/4 thrombocytopenia), while patients with advanced solid malignancies and previously treated sarcoma had much lower rates (15.9% and 9.3%, respectively) [56, 57]. Fortunately, given the short half-life of selinexor, drug interruption and dose reductions upon disease control are important strategies to reverse toxicities, in addition to proactive supportive care, including combinations of antiemetics. Moreover, similar to carfilzomib, pomalidomide, and daratumuab, the optimal use of novel agents, including selinexor, is not as a doublet derived from the accelerated approval studies, but rather triplet combinations to overcome the genomic and immunologic complexity of heavily pretreated MM. The choice of the third agent is based on efficacy, toxicity, and approval/availability. Such triplet combinations also allow weekly dosing of selinexor at a dose lower than that needed in the STORM study, with attendant reduction in toxicity. Attention must also be given to monitoring for corticosteroid toxicities (e.g. cataracts) and making appropriate dose adjustments.
Eventually, as more trials are completed, we may see selinexor moved to earlier lines of treatment where patients may tolerate the agent better due to better functional status and lower adverse effect burden from prior therapies. Based on preliminary data from the BOSTON study, this may be a consideration, especially for those patients with high-risk MM (e.g. 17p deletion). The next-generation SINE eltanexor, with a similar mechanism to selinexor, has been shown to have a potentially improved adverse effect profile with similar efficacy, although more clinical data are needed at this time.
Given the recent accelerated approval in June 2020 for selinexor in DLBCL after at least two lines of systemic therapy based on the SADAL trial [58], as well as promising results from the phase II KING study (NCT01986348) in recurrent glioblastoma [59], it is likely that clinicians will become more familiar with managing toxicities and maximizing the therapeutic potential of this class of drugs. The results of ongoing clinical trials for selinexor and eltanexor are eagerly anticipated and will help further define the role of SINEs in MM and other malignancies.
References
Alanazi F, Kwa F, Burchall G, Jackson DE. New generation drugs for treatment of multiple myeloma. Drug Discov Today. 2020;25(2):367–79.
Lonial S, Dimopoulos M, Palumbo A, White D, Grosicki S, Spicka I, et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N Engl J Med. 2015;373(7):621–31.
Gandhi UH, Cornell R, Lakshman A, Gahvari Z, McGehee E, Jagosky M, et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia. 2019;33(9):2266–75.
Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol. 2020;83(8):1021–32.
Huang ZL, Gao M, Li QY, Tao K, Xiao Q, Caso WX, et al. Induction of apoptosis by directing oncogenic Bcr-Abl into the nucleus. Oncotarget. 2013;4(12):2249–60.
Culjkovic-Kraljacic B, Baguet A, Volpon L, Amri A, Borden K. The oncogene eIF4E reprograms the nuclear pore complex to promote mRNA export and oncogenic transformation. Cell Rep. 2012;2(2):207–15.
Zhou F, Qiu W, Yao R, Xiang J, Sun X, Liu S, et al. CRM1 is a novel independent prognostic factor for the poor prognosis of gastric carcinomas. Med Oncol. 2013;30(4):726.
Kojima K, Kornblau S, Ruvolo V, Dilip A, Dubburi S, Davis R, et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood. 2013;121(20):4166–74.
Gandhi UH, Senapedis W, Baloglu E, Unger T, Chari A, Vagl D, et al. Clinical implications of targeting XPO1-mediated nuclear export in multiple myeloma. Clin Lymph Myeloma Leuk. 2018;18(5):335–45.
Schmidt J, Braggio E, Kortuem KM, Egan JB, Zhu YX, Xin CS, et al. Genome-wide studies in multiple myeloma identify XPO1/CRM1 as a critical target validated using the selective nuclear export inhibitor KPT-276. Leukemia. 2013;27(12):2357–65.
Camus V, Miloudi H, Taly A, Sola B, Jardin F. XPO1 in B cell hematological malignancies: from recurrent somatic mutations to targeted therapy. J Hematol Oncol. 2017;10(1):47.
Chanukuppa V, Paul D, Taunk K, Chatterjee T, Sharma S, Kumar S, et al. XPO1 is a critical player for bortezomib resistance in multiple myeloma: a quantitative proteomic approach. J Proteom. 2019;209:103504.
US FDA. FDA grants accelerated approval to selinexor for multiple myeloma. 2019. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-selinexor-multiple-myeloma. Accessed 15 Sept 2020.
Hamamoto T, Seto H, Beppu T. Leptomycins A and B, new antifungal antibiotics II. Structure elucidation. J Antibiot (Tokyo). 1983;36(6):646–50.
Nishi K, Yoshida M, Fujiwara D, Nishikawa M, Horinouchi S, Beppu T. Leptomycin B targets a regulatory cascade of crm1, a fission yeast nuclear protein, involved in control of higher order chromosome structure and gene expression. J Biol Chem. 1994;269(9):6320–4.
Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner E, Wolff B, et al. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci USA. 1999;96(16):9112–7.
Roberts BJ, Hamelehle KL, Sebolt JS, Leopold WR. In vivo and in vitro anticancer activity of the structurally novel and highly potent antibiotic CI-940 and its hydroxy analog (PD 114,721). Cancer Chemother Pharmacol. 1986;16(2):95–101.
Newlands ES, Rustin GJ, Brampton MH. Phase I trial of elactocin. Br J Cancer. 1996;74(4):648–9.
Burzlaff A, Kalesse M, Kasper C, Scheper T. Multi parameter in vitro testing of ratjadone using flow cytometry. Appl Microbiol Biotechnol. 2003;62(2–3):174–9.
Turner JG, Marchion DC, Dawson JL, Emmons MF, Hazlehurst LA, Washausen P, et al. Human multiple myeloma cells are sensitized to topoisomerase II inhibitors by CRM1 inhibition. Cancer Res. 2009;69(17):6899–905.
Hatcher H, Planalp R, Cho J, Torti F, Torti S. Curcumin: from ancient medicine to current clinical trials. Cell Mol Life Sci. 2008;65(11):1631–52.
Mingshan N, Sinjin W, Yongliag Y. CRM1 is a cellular target of curcumin: new insights for the myriad of biological effects of an ancient spice. Traffic. 2013;14(10):1042–52.
Lee EW, Oh W, Song HP, Kim WK. Phosphorylation of p53 at threonine 155 is required for Jab1-mediated nuclear export of p53. BMB Rep. 2017;50(7):373–8.
Gomez-Bougie P, Halliez M, Maïga S, Godon C, Kervoelen C, Pellat-Deceunynck C, et al. Curcumin induces cell death of the main molecular myeloma subtypes, particularly the poor prognosis subgroups. Cancer Biol Ther. 2015;16(1):60–5.
Golombick T, Diamond T, Manoharan A, Ramakrishna R. Monoclonal gammopathy of undetermined significance, smoldering multiple myeloma, and curcumin: a randomized, double-blind placebo-controlled cross-over 4g study and an open-label 8g extension study. Am J Hematol. 2012;87(5):455–60.
Ramakrishna R, Diamond T, Alexander W, Manoharan A, Golombick T. Use of curcumin in multiple myeloma patients intolerant of steroid therapy. Clin Case Rep. 2020;8(4):739–44.
Bolger G, Licollari A, Bagshaw R, Tan A, Greil R, Vcelar B, et al. Intense uptake of liposomal curcumin by multiple myeloma cell lines: comparison to normal lymphocytes, red blood cells and chronic lymphocytic leukemia cells. Anticancer Res. 2019;39(3):1161–8.
Greil R, Greil-Ressler S, Weiss L, Schonlieb C, Magnes T, Radl B, et al. A phase 1 dose-escalation study on the safety, tolerability and activity of liposomal curcumin (LipocurcTM) in patients with locally advanced or metastatic cancer. Cancer Chemother Pharmacol. 2018;82(4):695–706.
Sakakibara K, Saito N, Sato T, Suzuki A, Hasegawa Y, Friedman J, et al. CBS9106 is a novel reversible oral CRM1 inhibitor with CRM1 degrading activity. Blood. 2011;118(14):3922–31.
Wang J, Barve M, Chiorean E, LoRusso P, Courtney K, Qi D, et al. Interim results from trial of SL-801, a novel XPO-1 inhibitor, in patients with advanced solid tumours. Ann Oncol. 2019;30(Suppl 5):v175.
Kalid O, Warshaviak DT, Shechter S, Sherman W, Shacham S. Consensus Induced Fit Docking (cIFD): methodology, validation, and application to the discovery of novel Crm1 inhibitors. J Comput Aided Mol Des. 2020;26(11):1217–28.
Parikh K, Cang S, Sekhri A, Liu D. Selective inhibitors of nuclear export (SINE)–a novel class of anti-cancer agents. J Hematol Oncol. 2014;15(7):78.
Sendino M, Omaetxebarria M, Rodriguez J. Hitting a moving target: inhibition of the nuclear export receptor XPO1/CRM1 as a therapeutic approach in cancer. Cancer Drug Resist. 2018;1:139–63.
Turner JG, Dawson J, Emmons MF, Cubitt CL, Kauffman M, Shacham S, et al. CRM1 inhibition sensitizes drug resistant human myeloma cells to topoisomerase II and proteasome inhibitors both in vitro and ex vivo. J Cancer. 2013;4(8):614–25.
Rosebeck S, Alonge M, Kandarpa M, Mayampurath A, Volchenboum S, Jasielec J, et al. Synergistic myeloma cell death via novel intracellular activation of caspase-10-dependent apoptosis by carfilzomib and selinexor. Mol Cancer Ther. 2016;15(1):60–71.
Turner JG, Dawson JL, Grant S, Shain K, Dalton W, Dai Y, et al. Treatment of acquired drug resistance in multiple myeloma by combination therapy with XPO1 and topoisomerase II inhibitors. J Hematol Oncol. 2016;9(1):73.
Turner JG, Kashyap T, Dawson JL, Gomez J, Bauer A, Grant S, et al. XPO1 inhibitor combination therapy with bortezomib or carfilzomib induces nuclear localization of IκBα and overcomes acquired proteasome inhibitor resistance in human multiple myeloma. Oncotarget. 2016;7(48):78896–909.
Muz B, Azab F, de la Puente P, Landesman Y, Zab AK. Selinexor overcomes hypoxia-induced drug resistance in multiple myeloma. Transl Oncol. 2017;10(4):632–40.
Tai YT, Landesman Y, Acharya C, Calle Y, Zhong MY, Cea M, et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications. Leukemia. 2014;28(1):155–65.
Argueta C, Kashyap T, Klebanov B, Unger T, Guo C, Harrington S, et al. Selinexor synergizes with dexamethasone to repress mTORC1 signaling and induce multiple myeloma cell death. Oncotarget. 2018;9(39):25529–44.
Chari A, Vogl D, Gavriatopoulou M, Nooka A, Yee A, Huff C, et al. Oral selinexor-dexamethasone for triple-class refractory multiple myeloma. N Engl J Med. 2019;381(8):727–38.
Aapro MS, Walko CM. Aprepitant: drug-drug interactions in perspective. Ann Oncol. 2010;21(12):2316–23.
Dimopoulos M, Delimpasi S, Simonova M, Spicka I, Ludek P, Kryachok I, et al. Weekly selinexor, bortezomib, and dexamethasone (SVd) versus twice weekly bortezomib and dexamethasone (Vd) in patients with multiple myeloma (MM) after one to three prior therapies: initial results of the phase III BOSTON study. J Clin Oncol. 2020;38(Suppl 15):8501.
Chari A, Vogl D, Jagannath S, Jasielec J, Unger T, DeCastro A, et al. Selinexor-based regimens for the treatment of myeloma refractory to chimeric antigen receptor T cell therapy. Br J Haematol. 2020;189:126–30.
Jakubowiak AJ, Jasielec J, Rosenbaum CA, Cole CE, Chari A, Mikael J, et al. Phase 1 study of selinexor plus carfilzomib and dexamethasone for the treatment of relapsed/refractory multiple myeloma. Br J Haematol. 2019;186(4):549–60.
Chen C, Gasparetto C, White D, Kotb R, Lipe B, Sutherland H, et al. Selinexor, pomalidomide, and dexamethaspone (SPD) in patients with relapsed or refractory multiple myeloma. EHA 24 Abstract PF587. 2019.
Jei J. Antegene Corporation Treating Patients Without Borders. Business Confidential. 2019. https://www.jefferies.com/CMSFiles/Jefferies.com/files/Antengene%20Corporation%20v3.pdf. Accessed 15 Sept 2020.
White D, LeBlanc R, Venner C, Bahlis N, Lentzsch S, Gasparetto C, et al. Safety and efficacy of the combination of selinexor, lenalidomide and dexamethasone (SRd) in patients with relapsed/refractory multiple myeloma (RRMM) [abstract no. 3532019]. In: Presented at the 17th International Myeloma Workshop; 12–15 Sep 2019: Boston, MA.
Gasparetto C, Lentzsch S, Gary J, Callander N, Tuchman S, Bahlis N, et al. Selinexor, daratumumab, and dexamethasone in patients with relapsed/refractory multiple myeloma (MM). J Clin Oncol. 2020;38(Suppl 15):8510.
Gasparetto C, Lipe B, Tuchman S, Callander N, Lentzsch S, Baljevic M, et al. Once weekly selinexor, carfilzomib, and dexamethasone (SKd) in patients with relapsed/refractory multiple myeloma (MM). J Clin Oncol. 2020;38(Suppl 15):8530.
Hing ZA, Fung HY, Ranganathan P, Mitchell S, El-Gamal D, Woyach JA, et al. Next-generation XPO1 inhibitor shows improved efficacy and in vivo tolerability in hematological malignancies. Leukemia. 2016;30(12):2364–72.
Turney J, Dawson J, Cubitt C, Baluglo E, Grant S, Dai Y, et al. Next generation XPO1 inhibitor KPT-8602 for the treatment of drug-resistant multiple myeloma. Blood. 2015;126(23):1818.
Karyopharm Presents Positive Phase 1/2 Eltanexor Data at the American Society of Hematology 2017 Annual Meeting [news release]. 2017. https://investors.karyopharm.com/news-releases/news-release-details/karyopharm-presents-positive-phase-12-eltanexor-data-american. Accessed 15 Sept 2020.
Rajkumar SV. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am J Hematol. 2020;95(5):548–67.
Kortuem K, Zidich K, Schuster S, Khan M, Jimenez-Zepeda V, Mikhael J, et al. Activity of 129 single-agent drugs in 228 phase I and II clinical trials in multiple myeloma. Clin Lymph Myeloma Leuk. 2014;14(4):284–90.
Gounder M, Zer A, Tap W, Salah S, Dickson M, Gupta A, et al. Phase IB study of selinexor, a first-in-class inhibitor of nuclear export, in patients with advanced refractory bone or soft tissue sarcoma. J Clin Oncol. 2016;34(26):3166–74.
Abdul Razak AR, Mau-Soerensen M, Gabrail NY, et al. First-in-class, first-in-human phase I study of selinexor, a selective inhibitor of nuclear export, in patients with advanced solid tumors. J Clin Oncol. 2016;34(34):4142–50.
US FDA. FDA approves selinexor for relapsed/refractory diffuse large B-cell lymphoma. 2020. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-selinexor-relapsedrefractory-diffuse-large-b-cell-lymphoma. Accessed 15 Sept 2020.
Lassman A, Wen P, Bent V, Plotkin S, Walenkamp A, Huang X, et al. Efficacy and safety of selinexor in recurrent glioblastoma. J Clin Oncol. 2019;37(Suppl 15):2005.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
None.
Conflicts of interest/Competing interests
Nicholas Theodoropoulos and Guido Lancman declare they have no conflicts of interest. Ajai Chari reports research support from Janssen, Celgene, Novartis Pharmaceuticals, Amgen, Pharmacyclics, Seattle Genetics, and Millenium/Takeda, and also reports consulting fees from Janssen, Celgene, Novartis Pharmaceuticals, Amgen, BMS, Karyopharm, Sanofi Genzyme, Seattle Genetics, Oncopeptides, Millenium/Takeda, Antengene, GlaxoSmithKline, and Secura Bio.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Rights and permissions
About this article

Cite this article
Theodoropoulos, N., Lancman, G. & Chari, A. Targeting Nuclear Export Proteins in Multiple Myeloma Therapy. Targ Oncol 15, 697–708 (2020). https://doi.org/10.1007/s11523-020-00758-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-020-00758-2