
Abstract
Inherited syndromes of intrahepatic cholestasis and biliary atresia are the most common causes of chronic liver disease and the prime indication for liver transplantation in children. Our understanding of the pathogenesis of these diseases has increased substantially by the discovery of genetic mutations in children with intrahepatic cholestasis and the findings that inflammatory circuits are operative at the time of diagnosis of biliary atresia. Building on this solid foundation, recent studies provide new insight into genotype-phenotype relationships and how mutations produce altered bile composition and cholestasis. New evidence exists that although liver transplantation is curative for patients with end-stage liver disease owing to cholestasis, some patients may develop recurrence of cholestasis because of the emergence of autoantibodies that disrupt canalicular function in the new graft. Progress is also evident in biliary atresia, with recent studies identifying candidate modifier genes and directly implicating lymphocytes and inflammatory signals in the pathogenesis of bile duct injury and obstruction.


Similar content being viewed by others

References
Papers of particular interest, published recently, have been highlighted as: • Of importance•• Of major importance
Balistreri WF, Heubi JE, Suchy FJ: Immaturity of the enterohepatic circulation in early life: factors predisposing to “physiologic” maldigestion and cholestasis. J Pediatr Gastroenterol Nutr 1983, 2:346–354.
Balistreri WF, Bezerra JA: Whatever happened to "neonatal hepatitis"? Clin Liver Dis 2006, 10:27–53.
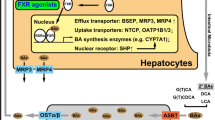
Wagner M, Zollner G, Trauner M: New molecular insights into the mechanisms of cholestasis. J Hepatol 2009, 51:565–580.
Strople J, Lovell G, Heubi J: Prevalence of subclinical vitamin k deficiency in cholestatic liver disease. J Pediatr Gastroenterol Nutr 2009, 49:78–84.
Whitington PF, Hibbard JU: High-dose immunoglobulin during pregnancy for recurrent neonatal haemochromatosis. Lancet 2004, 364:1690–1698.
•• Whitington PF, Kelly S: Outcome of pregnancies at risk for neonatal hemochromatosis is improved by treatment with high-dose intravenous immunoglobulin. Pediatrics 2008, 121:e1615–e1621. This article reports the impact of IV Ig administered during pregnancy on the outcome of neonates with iron storage disease. IV Ig was administered weekly to women from 18 weeks until the end of at-risk pregnancies. Gestational therapy reduced the recurrence rate of liver disease and promoted survival in 98% of neonates.
Pan S, Huang L, McPherson J, et al.: Single nucleotide polymorphism–mediated translational suppression of endoplasmic reticulum mannosidase I modifies the onset of end-stage liver disease in alpha1-antitrypsin deficiency. Hepatology 2009, 50:275–281.
• Bochkis IM, Rubins NE, White P, et al.: Hepatocyte-specific ablation of Foxa2 alters bile acid homeostasis and results in endoplasmic reticulum stress. Nat Med 2008, 14:828–836. This report describes the biologic consequences of the inactivation of the Foxa2 gene in mice. Loss of Foxa2 resulted in intrahepatic cholestasis associated with a decreased expression of genes involved in bile acid transport at the basolateral and canalicular membranes. Foxa2 was also found to be decreased in children and adults with cholestatic syndromes.
• Miyagawa-Hayashino A, Egawa H, Yorifuji T, Hasegawa M, et al.: Allograft steatohepatitis in progressive familial intrahepatic cholestasis type 1 after living donor liver transplantation. Liver Transpl 2009, 15:610–618. This article reports the development of macrovesicular steatosis in 8 of 11 patients who received living-donor liver transplantation for progressive familial intrahepatic cholestasis. Patients developed steatohepatitis (7 of 8), bridging fibrosis (6 of 8), or cirrhosis (2 of 8) in the liver graft. Posttransplant diarrhea occurred in all eight patients with hepatic steatosis.
Paulusma CC, Folmer DE, Ho-Mok KS, et al.: ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology 2008, 47:268–278.
Folmer DE, van der Mark VA, Ho-Mok KS, et al.: Differential effects of progressive familial intrahepatic cholestasis type 1 and benign recurrent intrahepatic cholestasis type 1 mutations on canalicular localization of ATP8B1. Hepatology 2009, 50:1597–1605.
• Frankenberg T, Miloh T, Chen FY, et al.: The membrane protein ATPase class I type 8B member 1 signals through protein kinase C zeta to activate the farnesoid X receptor. Hepatology 2008, 48:1896–1905. This article reports the functional relationship between ATP8B1 (FIC1), FXR, and BSEP promoter using a cell-culture system. The investigators found that the overexpression of FIC1 in a cell culture system promoted phosphorylation and nuclear localization of FXR, which was blocked by protein kinase C zeta inhibitors. By expressing FIC1 mutants based on sequence results from patients with PFIC-1 and BRIC-1, they found that severe mutations in FIC1 were unable to induce FXR-dependent activation of BSEP, whereas milder mutations (ie, from patients with BRIC-1) partially activated BSEP.
• Cai SY, Gautam S, Nguyen T, et al.: ATP8B1 deficiency disrupts the bile canalicular membrane bilayer structure in hepatocytes, but FXR expression and activity are maintained. Gastroenterology 2009, 136:1060–1069. This article investigated whether ATP8B1 deficiency produces cholestasis by affecting FXR activity or by impairing the structure of the canalicular membrane. Knocking down the ATP8B1 gene using a siRNA-based approach, the authors found no changes in the expression of FXR or FXR-dependent membrane transporters. In contrast, cells with suppressed ATP8B1 had focal areas of canalicular disruption by electron microscopy when exposed to bile acids.
Wang L, Dong H, Soroka CJ, et al.: Degradation of the bile salt export pump at endoplasmic reticulum in progressive familial intrahepatic cholestasis type II. Hepatology 2008, 48:1558–1569.
Byrne JA, Strautnieks SS, Ihrke G, et al.: Missense mutations and single nucleotide polymorphisms in ABCB11 impair bile salt export pump processing and function or disrupt pre-messenger RNA splicing. Hepatology 2009, 49:553–567.
•• Strautnieks SS, Byrne JA, Pawlikowska L, et al.: Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology 2008, 134:1203–1214. This article describes the clinical phenotype and biallelic mutations in most children diagnosed with deficiency of BSEP. About 93% of the subjects had abnormal or absent BSEP on liver biopsies. Fifteen percent of the patients also developed hepatocellular carcinoma/cholangiocarcinoma, with the incidence further increasing to 38% if the mutations produced truncated proteins.
Knisely AS, Strautnieks SS, Meier Y, et al.: Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology 2006, 44:478–486.
Scheimann AO, Strautnieks SS, Knisely AS, et al.: Mutations in bile salt export pump (ABCB11) in two children with progressive familial intrahepatic cholestasis and cholangiocarcinoma. J Pediatr 2007, 150:556–559.
•• Keitel V, Burdelski M, Vojnisek Z, et al.: De novo bile salt transporter antibodies as a possible cause of recurrent graft failure after liver transplantation: a novel mechanism of cholestasis. Hepatology 2009, 50:510–517. This article reports the clinical features and outcome of a child who developed recurrence of cholestatic liver disease following transplantation for complications of BSEP deficiency. The patient’s serum was reactive to a domain of the extracellular loop of BSEP and inhibited its transport activity.
•• Jara P, Hierro L, Martinez-Fernandez P, et al.: Recurrence of bile salt export pump deficiency after liver transplantation. N Engl J Med 2009, 361:1359–1367. This article reports the clinical features and outcome of three children who developed recurrence of cholestatic liver disease following transplantation for end-stage liver disease due to BSEP deficiency. All patients had circulating high-titer antibodies that recognized BSEP and inhibited its transport properties.
Delaunay JL, Durand-Schneider AM, Delautier D, et al.: A missense mutation in ABCB4 gene involved in progressive familial intrahepatic cholestasis type 3 leads to a folding defect that can be rescued by low temperature. Hepatology 2009, 49:1218–1227.
de Vree JM, Jacquemin E, Sturm E, et al.: Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A 1998, 95:282–287.
Deleuze JF, Jacquemin E, Dubuisson C, et al.: Defect of multidrug-resistance 3 gene expression in a subtype of progressive familial intrahepatic cholestasis. Hepatology 1996, 23:904–908.
Trauner M, Fickert P, Wagner M: MDR3 (ABCB4) defects: a paradigm for the genetics of adult cholestatic syndromes. Semin Liver Dis 2007, 27:77–98.
• Ziol M, Barbu V, Rosmorduc O, et al.: ABCB4 heterozygous gene mutations associated with fibrosing cholestatic liver disease in adults. Gastroenterology 2008, 135:131–141. This article reports the mutation analysis of the ABCB4 gene in 32 adults with anicteric cholestasis of unknown etiology. The authors found heterozygous mutations in 34% of patients, with decreased or absent MDR3 staining.
Makin E, Quaglia A, Kvist N, et al.: Congenital biliary atresia: liver injury begins at birth. J Pediatr Surg 2009, 44:630–633.
Serinet MO, Wildhaber BE, Broue P, et al.: Impact of age at Kasai operation on its results in late childhood and adolescence: a rational basis for biliary atresia screening. Pediatrics 2009, 123:1280–1286.
Hsiao CH, Chang MH, Chen HL, et al.: Universal screening for biliary atresia using an infant stool color card in Taiwan. Hepatology 2008, 47:1233–1240.
Kieling CO, Santos JL, Vieira SM, et al.: Biliary atresia: we still operate too late. J Pediatr (Rio J) 2008, 84:436–441.
Sokol RJ, Shepherd RW, Superina R, et al.: Screening and outcomes in biliary atresia: summary of a National Institutes of Health workshop. Hepatology 2007, 46:566–581.
Ohhama Y, Shinkai M, Fujita S, et al.: Early prediction of long-term survival and the timing of liver transplantation after the Kasai operation. J Pediatr Surg 2000, 35:1031–1034.
Shneider BL, Brown MB, Haber B, et al.: A multicenter study of the outcome of biliary atresia in the United States, 1997 to 2000. J Pediatr 2006, 148:467–474.
Pape L, Olsson K, Petersen C, et al.: Prognostic value of computerized quantification of liver fibrosis in children with biliary atresia. Liver Transpl 2009, 15:876–882.
Santos JL, Kieling CO, Meurer L, et al.: The extent of biliary proliferation in liver biopsies from patients with biliary atresia at portoenterostomy is associated with the postoperative prognosis. J Pediatr Surg 2009, 44:695–701.
Bezerra JA: The next challenge in pediatric cholestasis: deciphering the pathogenesis of biliary atresia. J Pediatr Gastroenterol Nutr 2006, 43(Suppl 1):S23–S29.
Yokoyama T, Copeland NG, Jenkins NA, et al.: Reversal of left-right asymmetry: a situs inversus mutation. Science 1993, 260:679–682.
Davit-Spraul A, Baussan C, Hermeziu B, et al.: CFC1 gene involvement in biliary atresia with polysplenia syndrome. J Pediatr Gastroenterol Nutr 2008, 46:111–112.
Lee HC, Chang TY, Yeung CY, et al.: Genetic variation in the vascular endothelial growth factor gene is associated with biliary atresia. J Clin Gastroenterol 2009 (Epub ahead of print).
• Spence JR, Lange AW, Lin SC, et al.: Sox17 regulates organ lineage segregation of ventral foregut progenitor cells. Dev Cell 2009, 17:62–74. This article reports the anatomic consequences of under-and overexpression of Sox17, a gene involved in determination of endoderm lineage. Loss of Sox17 in mice resulted in the absence of biliary structures, while the overexpression suppressed pancreatic development and facilitated the expansion of ectopic biliary-like tissue.
• Yamashita R, Takegawa Y, Sakumoto M, et al.: Defective development of the gallbladder and cystic duct in Lgr4-hypomorphic mice. Dev Dyn 2009, 238:993–1000. This article reports the absence of the gallbladder and cystic duct in mice carrying the targeted inactivation of the Lgr4 gene. The remainder of the extrahepatic bile ducts was normal.
Kobayashi H, Tamatani T, Tamura T, et al.: Maternal microchimerism in biliary atresia. J Pediatr Surg 2007, 42:987–991; discussion 991.
Suskind DL, Rosenthal P, Heyman MB, et al.: Maternal microchimerism in the livers of patients with biliary atresia. BMC Gastroenterol 2004, 4:14.
Muraji T, Hosaka N, Irie N, et al.: Maternal microchimerism in underlying pathogenesis of biliary atresia: quantification and phenotypes of maternal cells in the liver. Pediatrics 2008, 121:517–521.
Diaz R, Kim JW, Hui JJ, et al.: Evidence for the epithelial to mesenchymal transition in biliary atresia fibrosis. Hum Pathol 2008, 39:102–115.
Harada K, Sato Y, Ikeda H, et al.: Epithelial-mesenchymal transition induced by biliary innate immunity contributes to the sclerosing cholangiopathy of biliary atresia. J Pathol 2009, 217:654–664.
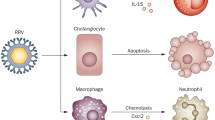
•• Jafri M, Donnelly B, Allen S, et al.: Cholangiocyte expression of alpha2beta1-integrin confers susceptibility to rotavirus-induced experimental biliary atresia. Am J Physiol Gastrointest Liver Physiol 2008, 295:G16–G26. This article reports the increased expression of α2β1-integrin in cholangiocytes using cell culture and in vivo assays. The expression of this integrin was important for the susceptibility of cholangiocytes to rotavirus infection. Blocking of the integrin using specific antibodies minimized symptoms and improved survival in an experimental model of rotavirus-induced biliary atresia.
• Barnes BH, Tucker RM, Wehrmann F, et al.: Cholangiocytes as immune modulators in rotavirus-induced murine biliary atresia. Liver Int 2009, 29:1253–1261. This article describes the expression of markers of antigen-presenting cells (MHC-I and II, CD40) in cholangiocyte cell lines and freshly isolated cells. However, cultured cholangiocytes were unable to function as competent antigen-presenting cells in T-cell proliferation assays.
•• Shivakumar P, Sabla GE, Whitington P, et al.: Neonatal NK cells target the mouse duct epithelium via Nkg2d and drive tissue-specific injury in experimental biliary atresia. J Clin Invest 2009, 119:2281–2290. The investigators analyzed the population of mononuclear cells in extrahepatic bile ducts of neonatal mice and found that NK cells are the most abundant cells. Using a rotavirus-induced model of biliary atresia, they also found an increase in number and activation status of NK cells, which used the Nkg2d receptor to attach to and kill cholangiocytes. The loss of NK cells or blocking of Nkg2d using antibodies prevented injury to the epithelium of extrahepatic bile ducts, decreased symptoms, and promoted long-term survival of neonatal mice challenged with rotavirus.
•• Davenport M, Stringer MD, Tizzard SA, et al.: Randomized, double-blind, placebo-controlled trial of corticosteroids after Kasai portoenterostomy for biliary atresia. Hepatology 2007, 46:1821–1827. This article describes the first prospective, randomized, double-blind trial of corticosteroids following portoenterostomy in infants with biliary atresia. The investigators found no difference in biliary flow or transplant-free survival between infants treated with corticosteroids and those receiving placebo, except for improvement in serum bilirubin when corticosteroids were administered to infants younger than 70 days of age at the time of portoenterostomy.
Chung HY, Kak Yuen Wong K, Cheun Leung Lan L, et al.: Evaluation of a standardized protocol in the use of steroids after Kasai operation. Pediatr Surg Int 2008, 24:1001–1004.
Petersen C, Harder D, Melter M, et al.: Postoperative high-dose steroids do not improve mid-term survival with native liver in biliary atresia. Am J Gastroenterol 2008, 103:712–719.
Willot S, Uhlen S, Michaud L, et al.: Effect of ursodeoxycholic acid on liver function in children after successful surgery for biliary atresia. Pediatrics 2008, 122:e1236–e1241.
Disclosure
No potential conflict of interest relevant to this article was reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Santos, J.L., Choquette, M. & Bezerra, J.A. Cholestatic Liver Disease in Children. Curr Gastroenterol Rep 12, 30–39 (2010). https://doi.org/10.1007/s11894-009-0081-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11894-009-0081-8