
Abstract
A decade has passed since the discovery of angiotensin-converting enzyme 2 (ACE2), a component of the ACE2–angiotensin (Ang)-(1-7)–Mas counterregulatory axis of the renin angiotensin system (RAS). ACE2 is considered an endogenous regulator of the vasoconstrictive, proliferative, fibrotic, and proinflammatory effects of the ACE–Ang II–angiotensin II type 1 receptor (AT1R) axis. Both animal and clinical studies have emerged to define a role for ACE2 in pulmonary arterial hypertension (PAH). There is scientific evidence supporting the concept that ACE2 maintains the RAS balance and plays a protective role in PAH. The activation of pulmonary ACE2 could influence the pathogenesis of PAH and serve as a novel therapeutic target in PAH. Current therapeutic strategies and interventions have limited success, and PAH remains a fatal disease. Thus, more research that establishes the novel therapeutic potential and defines the mechanism of the ACE2–Ang-(1-7)–Mas counterregulatory axis in PAH is needed.
Similar content being viewed by others


Introduction
Angiotensin (Ang)-converting enzyme 2 (ACE2) has been implicated in a number of physiologic and pathophysiologic processes. It is a novel homologue of ACE and a component of the counterregulatory axis of ACE. It is a monocarboxypeptidase, and it generates Ang-(1-7), another component of the renin-angiotensin system (RAS), which attenuates the vasoconstrictive, proliferative, fibrotic, and inflammatory effects of Ang II [1]. ACE2 cleaves a single residue from its substrate, Ang I, generating Ang-(1-9) and a single residue from another preferred substrate, Ang II, to generate Ang-(1-7). ACE2 also cleaves other peptide substrates such as des-arg-bradykinin, neurotensin, and kinetensin. ACE2 plays a pertinent role in the vasoprotective axis of the RAS, ACE2–Ang-(1-7)–Mas, as it counterbalances the vasoconstrictive, proliferative, and fibrotic actions of the ACE–Ang II–Ang II type 1 receptor (AT1R) axis [2••].
ACE2 is abundantly expressed in many cell types in the lung, such as Clara cells, type I and II alveolar epithelial cells, macrophages, endothelium, smooth muscle cells (SMCs) of blood vessels, and bronchial epithelia [3]. Because the RAS components are widely expressed in the lung, the activation of pulmonary ACE2 could influence the pathogenesis of lung injury and serve as a novel therapeutic target in pulmonary arterial hypertension (PAH).
PAH is a chronic disease of diverse etiology. Despite modern therapeutic advances, the World Health Organization functional class (WHO-FC) estimates median survival to be 6 months for WHO-FC IV, 2.5 years for WHO-FC III, and 6 years for WHO-FC I/II. Although targeted treatment, pleiotropic drug approaches, and novel progenitor-cell therapy provide symptomatic relief and prolong survival, PAH remains a fatal disease with no cure. Without proper therapy, chronic vasoconstriction, inflammation, and in situ thrombosis promote increased pulmonary vascular remodeling (PVR). Clinically, PAH is characterized by increased mean pulmonary arterial pressure (>25 mm Hg at rest) and persistent elevation of pulmonary vascular resistance, which leads to the main cause of death in these patients, right-sided heart failure [4].
PAH can be heritable, and predisposing genetic and environmental risk factors may lead to an imbalance in counterregulatory mechanisms associated with pulmonary vascular remodeling, such as vasoconstriction/vasodilatation, proliferation/antiproliferation, and prothrombogenics/antithrombogenics. These imbalances initiate a cascade of pathophysiologic events in the lungs leading to PAH [5••]; the exact pathogenesis of the disease is still unknown. Based upon the concept of homeostatic imbalance and multifactorial pathobiology, the mainstay of PAH treatment includes vasodilators, anticoagulants, calcium channel blockers, and endothelin receptor antagonists.
The renin-angiotensin system (RAS) is a well-recognized player in endothelial dysfunction and vascular remodeling, but the precise involvement of this system’s members in the lung pathophysiology of PAH remains elusive. Thus, the discovery of ACE2 and the emerging counterregulatory concept of RAS is of great significance [4]. The ACE–Ang II–AT1R axis promotes vasoconstriction, proliferation, and fibrosis, whereas the ACE2–Ang-(1-7)–Mas axis protects lungs. The ACE2–Ang-(1-7)–Mas axis intrinsically induces vasoprotective actions by counterregulating the ACE–Ang II–AT1R axis. This review focuses on the vasoprotective axis of the RAS as a potential target for therapeutic intervention in PAH. A conceptual breakthrough is urgently needed to develop a novel strategy for therapeutic intervention and management of PAH. Research establishing a role for ACE2 in lung pathophysiology is emerging.
The Role of the RAS in Pulmonary Arterial Hypertension
PAH is a chronic disease of diverse etiology, clinically characterized by increased pulmonary vascular resistance. Table 1 outlines the current classification of all forms of pulmonary hypertension (PH) [6••]. Genetic mutations influence PAH development, and in the year 2000, mutations of the gene encoding bone morphogenetic protein receptor type II (BMPR-II), a transforming growth factor (TGF)-β superfamily receptor, were identified as the primary genetic mutation in some patients with PAH [7].
PAH patients are often characterized by remodeling of small distal pulmonary arteries resulting in medial hypertrophy, intimal proliferation, adventitial thickening with moderate inflammatory infiltrates, plexiform lesions, and thrombotic lesions. This remodeling causes increased pulmonary vascular resistance resulting in a mean pulmonary arterial pressure greater than 25 mm Hg at rest. Persistent elevation of pulmonary vascular resistance leads to right heart failure and death [6••]. PAH is a consequence of vascular effector imbalance, perturbations in the homeostasis of vasoconstrictors and vasodilators, growth inhibitors and mitogenic factors, and antithrombotic and prothrombotic factors [8]. As a result of these findings, vasodilator, anticoagulant, antiplatelet, anti-inflammatory, and vascular remodeling therapies have been used for PAH treatment. Before current intervention strategies, life expectancy for adults with idiopathic PAH was less than 3 years from diagnosis; for children, it was less than 10 months [9]. There are complex strategies for treating PAH patients that include both supportive and specific drug therapy. Specific drug therapy often falls into three categories: vasodilators, calcium channel blockers, and endothelin receptor antagonists. The latest clinical trials have strategically targeted remodeling, including the testing of antiproliferative drugs in advanced human PAH.
Both clinical and animal data suggest that intervention strategies that attenuate the actions of the ACE–Ang II–AT1R axis (i.e., angiotensin receptor blockers and/or ACE inhibitor therapy) and activate the ACE2–Ang-(1-7)–Mas axis (i.e., ACE2 activation or ACE2 viral transfer) are effective in attenuating and preventing maladaptive vascular remodeling associated with PAH. Because limiting AngII bioactivity remains the cornerstone of cardiovascular therapeutics, it is plausible that the same approach may be valid in pulmonary diseases [10]. Pulmonary vascular fibrosis, hypertrophy, and vascular smooth muscle migration are regressed with ACE2. Thus, its discovery and role in lung pathophysiology are of great importance.
ACE2 and Its Role in Lung Diseases
ACE2 was initially discovered as a receptor for severe acute respiratory syndrome (SARS) coronavirus and because of its sequence homology to ACE, the research community began to determine its localization and study its role in most model systems. It is a type I transmembrane protein, which contains a single metalloproteinase active site [3]. ACE2 has been localized to human heart, kidney, coronary arteries, arterioles, vasa vasorum, testis, gastrointestinal tract, and lung. ACE2 is abundantly expressed in many cell types in the lung such as Clara cells, type I and II alveolar epithelial cells, macrophages, endothelium, SMCs, and bronchial epithelia. ACE2 is localized to endothelial cells, and expression is enhanced in the walls of newly muscularized pulmonary arteries [11]. These data illustrate that ACE2, like ACE, is localized to the lung, the vasculature, and more specifically, to the endothelium, and thus exists as a potential therapeutic target. The literature also strongly supports a RAS imbalance in pulmonary disease and lung injury.
ACE2 in Lung Injury
ACE2 limits vasoproliferative, fibrotic, and hypertrophic actions of the RAS during lung injury. Both animal and human pulmonary studies reveal that a RAS imbalance may play an important role in pulmonary disease progression. In three different models of acute respiratory distress syndrome (ARDS), ACE2 knockout mice have severe lung disease, elevated serum and tissue Ang II levels, and increased collagen deposition. This loss of ACE in acute lung injury leads to leaky pulmonary blood vessels through AT1a receptor stimulation [12]. In a lung injury model induced by acid aspiration or sepsis, ACE2 deletion worsens the injury. Aortas from ACE2-deficient mice exhibit impaired endothelium-dependent vasodilation [10]. This RAS imbalance is further supported by literature revealing that severe lung failure on an ACE2 knockout background was rescued by ACE inactivation [12].
The RAS imbalance is also evident in animal models of PH. Bleomycin-induced PH and pulmonary fibrosis are associated with decreased ACE2 activity. ACE2 is upregulated in response to hypoxia and illustrates a protective effect in severe acute lung failure. Activation of a key transcriptional factor during hypoxia, \( {\hbox{HIF}} - {1_\alpha } \), increased ACE and decreased ACE2 [13]. Lenti-ACE2 tracheal injections prevent and partially reverse PH in a monocrotaline (MCT) model of PH [14].
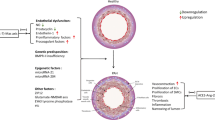
Targeting ACE2 may have additional beneficial effects, unlike the blockade of ACE and AT1Rs. Targeting ACE2 in PH is a novel strategy (Fig. 1), as ACE2 is a multifunctional enzyme with pleiotropic effects. It not only gives protection by degradation of Ang II, but it also produces the vasodilatory peptide, Ang-(1-7).

Targeting the ACE2/Ang-(1-7)/Mas axis prevents and reverses pulmonary vascular pathology of pulmonary arterial hypertension. ACE—angiotensin-converting enzyme; Ang II—angiotensin II; AT1R—angiotensin II type 1 receptor; AT2R—angiotensin II type 2 receptor; BMPR-II—bone morphogenetic protein receptor type II; ERK1/2—extracellular signal-regulated protein kinase 1/2; NO—nitric oxide; TGF-β—transforming growth factor β
In animal models, the literature also strongly supports pharmacologic treatment of lung disease with ACE2. Because PVR occurs in PAH, studies that prevent, limit, or regress PVR are important for progress in the field. In a sepsis-induced ACE2 knockout model of acute lung injury, treatment with recombinant human ACE2 protein provided direct protection [12]. This study also suggests that Ang II, ACE, and AT1a receptor promote lung injury, whereas ACE2 and AT2 receptor protect against lung injury. ACE2 overexpression improves endothelial cell migration and tube formation [10]. We have demonstrated in MCT-treated mice that pulmonary ACE2 overexpression via lentiviral vector both prevents and reverses the increases in right ventricular systolic pressure, significantly attenuates and partially reverses muscularization of pulmonary vessel, and increases the AT2R:AT1R mRNA ratio [14]. Our studies further reveal that activation of endogenous ACE2 by XNT (1-[(2-dimethylamino) ethylamino]-4-(hydroxymethyl)-7-[(4-methylphenyl) sulfonyl oxy]-9H-xanthene-9-one) prevents pulmonary vascular remodeling in PH development [2••]. XNT reversed cardiac hypertrophy, ventricular fibrosis, and renal interstitial fibrosis; prevented an increase in right ventricular systolic pressure (RVSP) and right ventricular hypertrophy; and attenuated vascular wall thickening and pulmonary fibrosis. XNT decreased proinflammatory cytokine levels, increased anti-inflammatory cytokine levels, and increased ACE2 activity. XNT had no adverse effects on systemic blood pressure. These effects are inhibited by an Ang-(1-7) antagonist, A779, demonstrating that ACE2 may produce beneficial effects via Ang-(1-7) [2••]. Interestingly, ACE2 overexpression in the smooth muscle of spontaneously hypertensive stroke prone rats (SHRSP) resulted in increased aortic ACE2 activity, Ang-(1-7), and Ang II and improved vascular function [15]. Mas agonists, AVE0991, CGEM856, CGEM857, confer endothelium-dependent relaxation effects.
Chronic and sustained imbalances that favor the ACE–Ang II–AT1R axis may promote the progression of lung disease. We believe that the beneficial effects of the system involve a shift from the vasoconstrictive, proliferative, and fibrotic axes to the vasoprotective axis of the RAS, and thus targeting the RAS in PH should be revisited, with ACE2 as the therapeutic target. Unlike ACE inhibitors and angiotensin-receptor blockers (ARBs), ACE2 is an endogenous regulator of the RAS. Therapy with ACE inhibitors and ARBs indirectly increases ACE2 and Ang-(1-7), but ACE2 activation would directly increase the enzyme to promote beneficial effects. Like ACE inhibitor and ARB therapy, ACE2 activation attenuates remodeling and fibrosis and affects the vascular relaxation properties of the endothelium. RAS intervention studies in PH are inconclusive, and the role of the ACE2–Ang-(1-7)–Mas axis activation in PH has not been confirmed with clinical trials.
RAS Intervention and Pulmonary Hypertension: The Controversy of ACE Inhibitor/ARB Therapy
It is evident that benchside pharmacologic inhibition of the RAS has translated to the bedside in the treatment of patients with hypertension, congestive heart failure, left ventricular dysfunction, pulmonary and systemic edema, diabetic nephropathy, cirrhosis of the liver, and scleroderma [16]. Although there is evidence in both clinical and animal studies supporting the contention that the RAS is involved in the development and progression of PAH, the use of ACE inhibitors in PAH remains controversial (Table 2).
RAS Therapy in Animal Models of Pulmonary Hypertension
Enalapril, another ACE inhibitor, was given concomitantly with MCT in a chronic PAH rat model fed a high-cholesterol diet [17]. This therapeutic intervention attenuated the development of PAH while preserving the expression of endothelial nitric oxide synthase (eNOS), demonstrating a role for RAS and NO in the development of PAH. In a hypoxic rat model of PAH, the dose-dependent effects of perindopril, an ACE inhibitor, on vascular remodeling did not restore pulmonary vascular function and had only a modest effect on pulmonary artery pressure [18]. Losartan, an AT1R antagonist, attenuated smoke-induced pulmonary artery remodeling, RVSP elevation, and Ang II accumulation, and increased ACE2 in rat lungs [19•]. Okada et al. [20] demonstrated that telmisartan, an AT1R antagonist, attenuated right ventricular (RV) remodeling via inhibition of RV hypertrophy, fibrosis, and dysfunction in MCT-treated male Wistar rats. Yet the acceleration time:ejection time ratio of pulmonary artery flow velocity was not significantly different than the ratio for controls. In clinical trials in human patients, ACE inhibitor therapy has been tested to treat PAH. Tavli et al. [21] conducted a small proof-of-concept trial in 30 PAH patients with stable chronic congestive heart failure and demonstrated that cilazapril, an ACE inhibitor, significantly decreased mean pulmonary and capillary wedge pressures. This drug also increased flow-mediated vasodilatation, improved left ventricular ejection fraction, and improved the functional class of patients. Both systemic and pulmonary arterial pressures were significantly decreased after 12 weeks of captopril treatment in 17 patients with high-altitude PH and mild-to-moderate systemic arterial hypertension [22]. Conversely, Bilan et al. [23] demonstrated in a 1-year placebo-controlled study of 41 PAH patients with scleroderma that enalapril did not alter LV systolic or diastolic function and heart diameter, and signs of PH were found in four of the patients. Zieliński et al. [24] demonstrated in 15 patients with hypoxic PH due to chronic obstructive lung disease that acute administration of captopril significantly decreased systemic arterial pressure but does not reduce PVR. In addition to blocking ACE and angiotensin receptors of the ACE–Ang II–AT1R axis, these inhibitors increase ACE2 and Ang-(1-7) expression levels in rats and humans [25]. In patients with primary PH, higher cardiac ACE2 activity and Ang-(1-7) levels provide cardioprotection; and in patients with pulmonary fibrosis, ACE2 expression is decreased [26]. These findings may indicate that ACE inhibition may be less effective in scleroderma and hypoxic PH or that it may be more effective in attenuation of remodeling when given earlier during the disease state. In addition to regressing remodeling, ACE inhibitors lower systemic blood pressure and therefore may not be beneficial for PH patients with right heart failure.
The ACE2–Ang-(1-7)–Mas RAS Axis: Mechanism of Beneficial Effects
There is a limited amount of evidence to support the mechanistic role of the ACE2–Ang-(1-7)–Mas axis. We do not completely understand how the ACE2–Ang-(1-7)–Mas axis protects the lungs from PH. Most of the literature has established that the Mas receptor is a G protein–coupled receptor, and its activation by Ang-(1-7) counteracts the actions of the ACE–Ang II–AT1R axis of the RAS system [27]. Yet the mechanisms involved are still unclear. Like ACE2 its downstream targets, Ang-(1-7) and Mas, demonstrate its impact on endothelial function. Mas knockout mice exhibited impaired endothelial function and decreased NO and eNOS [28]. In cardiac myocytes from Mas-deficient mice, Mas deficiency impairs Ang-(1-7) signaling [29].
In a recent article, brain-selective ACE2 overexpression attenuated neurogenic hypertension, possibly through Mas and AT2R upregulation [30]. Gallagher et al. [31•] reported that Ang-(1-7) prevented the Ang II–mediated reduction in ACE2 mRNA, and this was blocked by the Ang-(1-7) receptor antagonist [d-Ala7]-ANG-(1-7) and mitogen-activated protein (MAP) kinase kinase inhibitor PD98059. They were also the first to demonstrate in vascular SMCs that MAP kinase-phosphatase pathway regulates ACE2 and maintains the balance between Ang II and Ang-(1-7). Ang-(1-7) functions through NO or prostaglandins and has antifibrotic and antioxidant properties. Gwathmey et al. [32••] were the first to demonstrate that Ang-(1-7) promotes the proteolytic conversion of Ang II by ACE2 within the nucleus to potentially endogenously modulate reactive oxygen species (ROS) production. Wang et al. [33, 34] demonstrated that circulating Ang-(1-7) stimulated cardiac endothelial progenitor cell (EPC) proliferation benefiting myocardial infarction. Tallant et al. [35] suggest that Ang-(1-7) inhibits vascular growth through the release of prostacyclin, the prostacyclin-mediated production of cyclic adenosine monophosphate (cAMP), activation of cAMP-dependent protein kinase, and the attenuation of MAP kinase activation. These mechanistic studies have established that the ACE2–Ang-(1-7)–Mas receptor axis components exist within the nucleus and at every level of the vascular wall, poised to initiate and sustain a vasoprotective effect.
Signaling of TGF-β and Bone Morphogenetic Proteins in Pulmonary Hypertension
Ang II promotes growth, proliferation, and fibrosis. TGF-β upregulates ACE and downregulates ACE2. The TGF-β superfamily encompasses a large number of growth factors such as TGF-β ligands, activins, inhibins, and bone morphogenetic proteins (BMPs). BMPS are involved in vascular integrity and control of vascular cell proliferation [36]. Both TGF-β and BMP isoforms control endothelial cell and SMC proliferation, apoptosis, and extracellular matrix (ECM) secretion and deposition. BMPR-II and BMPR-1A plasma membrane protein expression is decreased in pulmonary endothelial cells in both heritable and sporadic PAH. BMPR-II has been localized to the endothelium and vascular SMCs of small pulmonary arteries [37]. Signal transduction studies reveal that BMP signals are mediated by type I and II receptors through downstream mediators, Smad1, Smad5, and Smad8. They form a complex with Smad4 and translocate into the nucleus. The function of BMPR-II is to mediate growth suppression and apoptosis in vasculature [38]. Thus a loss of BMPR-II function may lead to the vasculopathy evident in PAH [39]. Late cultured EPCs from idiopathic PAH patients with BMPR-II mutation showed a hyperproliferative phenotype with impaired ability to form vascular networks [40].
TGF-β signaling controls a number of functions: proliferation, migration, differentiation, apoptosis, ECM deposition, and secretion. Mutations in genes encoding either TGF-β or BMP signaling systems have been associated with PAH, and research suggests that abnormal TGF-β or BMP signaling is imbalanced in PAH [41]. Thus, TGF-β and BMP appear to play opposing roles (Fig. 1) in maintenance and growth of pulmonary artery SMCs and endothelial cells [42]. Studies suggest an inhibitory effect of BMP signaling on pulmonary artery SMCs. Such cells from PAH patients are resistant to antiproliferative effects of BMP. TGF-β gain of proliferation and BMPR-II loss of growth suppression and inability to promote vascular SMC apoptosis may promote PAH development.
Hong et al. [5••] revealed increased RVSP and increased muscularization in Bmpr2 knockout mice with incomplete penetrance. Reynolds et al. [43] demonstrated that overexpression of BMPR-II in a hypoxic rat model protects them against PAH development. BMPR-II heterozygous null mice have severe hypoxic PH with a decrease in eNOS expression and activity.
TGF-β signaling is modulated in MCT rat models and the results are controversial. Some studies suggest that MCT inhibits signaling, and others suggest that MCT enhances signaling. Inhibition of activin-like kinase 5 (ALK5), a TGF-β type I receptor, prevents and reverses PAH [44]. In both hypoxic and MCT models, BMPR-II mRNA and protein expression are reduced, with increased TGF-β and Smad3 signaling in the MCT model and not in the hypoxic model. This increase in signaling, RVSP, and vascular remodeling in the MCT model was attenuated by ALK5 inhibition [45]. Reduction in BMPR-II signaling due to genetic or environmental cues may cause BMPR-II signaling to fall below a threshold that triggers PAH vasculopathy. An imbalance in TGF–β/BMP signaling promotes PAH development and progression.
Future Directions
Since its discovery in 2000, ACE2 has been investigated in the SARS coronavirus infection and in the cardiovascular, renal, central nervous, and pulmonary systems. The benefit of its overexpression and activation in PAH supports its use in a “proof of concept” preclinical trial. It will be interesting to discover the link between TGF-β, BMPR-II, and the ACE2–Ang-(1-7)–Mas axis because, regardless of the disease etiology, remodeling consists of intimal fibrosis, increased medial thickness, pulmonary arteriolar occlusion, and plexiform lesions. ACE2 has been shown to prevent fibrosis and other components of PVR. Therefore, ACE2 therapy may be effective at preventing disease progression in all forms of PH.
Perturbation of BMP signaling is increased with missense mutations, and the knowledge of differences in disease severity and different BMPR-II mutations is critical to therapeutic design. Cilostazol, a phosphodiesterase III inhibitor, offers a synergistic benefit in ameliorating MCT-induced PAH in rats [46]. Rosuvastatin blunted the increase in RVSP but did not reduce mean arterial pressure in the Ren2 rat [47]. In a hypoxia-induced PH model, transplantation of Sca1+KDR+CXCR4+ cultured early EPCs failed to reverse vascular remodeling and changes in pulmonary hemodynamics, and intravenous administration of bone marrow cells failed to prevent an increase in RVSP and arterial muscularization, yet Raoul et al. [48] demonstrated that infusion of the same cells was effective in preventing MCT-induced PH. Zhao et al. [49] also showed that bone marrow–derived, endothelial-like progenitor cells were effective in preventing and reversing MCT-induced PH. Because pulmonary vascular remodeling encompasses: endothelial dysfunction, fibroblast and SMC activation, crosstalk between cells within the vascular wall, and recruitment of circulating progenitor cells, these may be potential therapeutic targets.
In addition to targeting activation or overexpression of ACE2, the literature illustrates other possible novel targets for intervention: phosphodiesterase inhibitors, statins, L-arginine, antiplatelet agents, serotonin inhibitors, agents to alter ion channel function, gene therapy, elastase inhibitors, antiproliferative heparins, tyrosine kinase inhibitors, and bone marrow–derived endothelial progenitor cell treatment [50].
The ACE–Ang II–AT1R axis of the RAS has been implicated in PAH, yet intervention studies using ACE inhibitors and ARBs remain controversial. Evidence targeting the ACE2–Ang-(1-7)–Mas axis of the RAS supports the idea that this axis antagonizes the proliferative, fibrotic, and hypertrophic effects of the ACE–Ang II–AT1R axis. In an effort to advance research efforts in PAH therapy, we must consider the efficacy of ACE2 activation in clinical trials, the use of subpressor doses of ACE inhibitors or ARBs in clinical trials, larger clinical trials, the efficacy of ACE inhibitors in regression of established lesions compared with new lesions, the timing of intervention, the identification of bone marrow cell types to treat PAH, and the development of animal models that reflect human PAH.
Conclusions
We need a novel strategic intervention, as PAH is a chronic, fatal disease. Current therapy addresses vasoconstrictor imbalance and endothelial dysfunction, but vasodilator therapy has yet to improve patient survival. Unlike epoprostenol, a prostacyclin analogue and potent vasodilator, most PAH therapies have not improved survival. Therefore, new therapies that target PVR are emerging. Increasing evidence supports the concept of an imbalance in various components of the RAS. Animal and human trials demonstrate that an imbalance that favors the ACE–Ang II–AT1R axis actions promotes proliferation, fibrosis, and growth. There is no current consensus for the efficacy of therapeutic PAH strategies such as ACEs or ARBs in human and animal models, yet the benefit of ACE2 overexpression and activation in lung is consistent. In PAH, ACE2 overexpression and activation exerts its effects via the ACE2–Ang-(1-7)–Mas axis, limiting the detrimental actions of the ACE–Ang II–AT1R axis and playing a protective role in PH. The exact mechanism of benefit remains elusive, but the benefit in both prevention and reversal studies in animal models is clear. Thus, activation of ACE2 is a novel and effective strategy for PAH that must be considered.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Grobe JL, Mecca AP, Lingis M, et al.: Prevention of angiotensin II-induced cardiac remodeling by angiotensin-(1-7). Am J Physiol Heart Circ Physiol 2007, 292:H736–H742.
•• Ferreira AJ, Shenoy V, Yamazato Y, et al.: Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension. Am J Respir Crit Care Med 2009, 179:1048–1054. XNT, an ACE2 activator, decreased renin, ACE, angiotensinogen, AT 1 R, and proinflammatory cytokines. It also prevented the development of PH and pulmonary vascular remodeling.
Hamming I, Cooper ME, Haagmans BL, et al.: The emerging role of ACE2 in physiology and disease. J Pathol 2007, 212:1–11.
Donoghue M, Hsieh F, Baronas E, et al.: A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res 2000, 87:E1–E9.
•• Hong KH, Lee YJ, Lee E, et al.: Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation 2008, 118:722–730. This study illustrates that pulmonary endothelial BMPRII deletion predisposes mice to PAH.
•• Galie N, Hoeper MM, Humbert M, et al.: Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009, 30:2493–2537. Experts in the field of pulmonary disease have provided a comprehensive review of published evidence for the management of PH. This includes the current classification scheme for all forms of PH.
Thomson JR, Machado RD, Pauciulo MW, et al.: Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-β family. J Med Genet 2000, 37:741–745.
Farber HW, Loscalzo J: Pulmonary arterial hypertension. N Engl J Med 2004, 351:1655–1665.
Runo JR, Loyd JE: Primary pulmonary hypertension. Lancet 2003, 361:1533–1544.
Lovren F, Pan Y, Quan A, et al.: Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am J Physiol Heart Circ Physiol 2008, 295:H1377–H1384.
Morrell NW, Morris KG, Stenmark KR: Role of angiotensin-converting enzyme and angiotensin II in development of hypoxic pulmonary hypertension. Am J Physiol 1995, 269:H1186–H1194.
Imai Y, Kuba K, Rao S, et al.: Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436:112–116.
Zhang R, Wu Y, Zhao M, et al.: Role of HIF-1alpha in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2009, 297:L631–L640.
Yamazato Y, Ferreira AJ, Hong KH, et al.: Prevention of pulmonary hypertension by angiotensin-converting enzyme 2 gene transfer. Hypertension 2009, 54:365–371.
Rentzsch B, Todiras M, Iliescu R, et al.: Transgenic angiotensin-converting enzyme 2 overexpression in vessels of SHRSP rats reduces blood pressure and improves endothelial function. Hypertension 2008, 52:967–973.
Harrison-Bernard LM: The renal renin-angiotensin system. Adv Physiol Educ 2009, 33:270–274.
Kanno S, Wu YJ, Lee PC, et al.: Angiotensin-converting enzyme inhibitor preserves p21 and endothelial nitric oxide synthase expression in monocrotaline-induced pulmonary arterial hypertension in rats. Circulation 2001, 104:945–950.
Jeffery TK, Wanstall JC: Perindopril, an angiotensin converting enzyme inhibitor, in pulmonary hypertensive rats: comparative effects on pulmonary vascular structure and function. Br J Pharmacol 1999, 128:1407–1418.
• Han SX, He GM, Wang T, et al.: Losartan attenuates chronic cigarette smoke exposure-induced pulmonary arterial hypertension in rats: possible involvement of angiotensin-converting enzyme-2. Toxicol Appl Pharmacol 2010, 245:100–107. Angiotensin receptor blockers inhibit ACE2 reduction in pulmonary artery smooth muscle cells in a model of smoke-induced PH.
Okada M, Harada T, Kikuzuki R, et al.: Effects of telmisartan on right ventricular remodeling induced by monocrotaline in rats. J Pharmacol Sci 2009, 111:193–200.
Tavli T, Gocer H: Effects of cilazapril on endothelial function and pulmonary hypertension in patients with congestive heart failure. Jpn Heart J 2002, 43:667–674.
Niazova ZA, Batyraliev TA, Aikimbaev KS, et al.: High-altitude pulmonary hypertension: effects of captopril on pulmonary and systemic arterial pressures. J Hum Hypertens 1996, 10(Suppl 3):S141–S142.
Bilan A, Chibowska M, Makaruk B, et al.: Enalapril (10 mg/day) in systemic sclerosis. One year, double blind, randomised study (ESS-1): echocardiographic substudy—three months follow-up. Adv Exp Med Biol 1999, 455:279–283.
Zielinski J, Hawrylkiewicz I, Gorecka D, et al.: Captopril effects on pulmonary and systemic hemodynamics in chronic cor pulmonale. Chest 1986, 90:562–565.
Chappell MC, Pirro NT, Sykes A, et al.: Metabolism of angiotensin-(1-7) by angiotensin-converting enzyme. Hypertension 1998, 31:362–367.
Li X, Molina-Molina M, Abdul-Hafez A, et al.: Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am J Physiol Lung Cell Mol Physiol 2008, 295:L178–L185.
Kostenis E, Milligan G, Christopoulos A, et al.: G-protein-coupled receptor Mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation 2005, 111:1806–1813.
Santo RA, Castro CH, Gava E, et al.: Impairment of in vitro and in vivo heart function in angiotensin-(1-7) receptor MAS knockout mice. Hypertension 2006, 47:996–1002.
Dias-Peixoto MF, Santos RA, Gomes ER, et al.: Molecular mechanisms involved in the angiotensin-(1-7)/Mas signaling pathway in cardiomyocytes. Hypertension 2008, 52:542–548.
Feng Y, Xia H, Cai Y, et al.: Brain-selective overexpression of human angiotensin-converting enzyme type 2 attenuates neurogenic hypertension. Circ Res 2010, 106:373–382.
• Gallagher PE, Ferrario CM, Tallant EA: MAP kinase/phosphatase pathway mediates the regulation of ACE2 by angiotensin peptides. Am J Physiol Cell Physiol 2008, 295:C1169–C1174. Gallagher et al. were the first to demonstrate in rat aortic vascular smooth muscle cells that the MAP kinase-phosphatase pathway regulates ACE2. AngII downregulated ACE2 activity and mRNA levels to maintain the AngII and Ang-(1-7) balance.
•• Gwathmey TM, Pendergrass KD, Reid SD, et al.: Angiotensin-(1-7)-angiotensin-converting enzyme 2 attenuates reactive oxygen species formation to angiotensin II within the cell nucleus. Hypertension 2010, 55:166–171. This was the first study to demonstrate a nuclear regulatory role of Ang-(1-7). In sheep kidney, it demonstrated that endogenous modulation of reactive oxygen species occurs by the preferential conversion of AngII to Ang-(1-7).
Wang LW, Fu XL, Clough R, et al.: Can angiotensin-converting enzyme inhibitors protect against symptomatic radiation pneumonitis? Radiat Res 2000, 153:405–410.
Wang Y, Qian C, Roks A, et al.: Circulating rather than cardiac angiotensin-(1-7) stimulates cardioprotection after myocardial infarction. Circ Heart Fail 2010, 3:286–293.
Tallant EA, Clark MA: Molecular mechanisms of inhibition of vascular growth by angiotensin-(1-7). Hypertension 2003, 42:574–579.
Fujiwara M, Yagi H, Matsuoka R, et al.: Implications of mutations of activin receptor-like kinase 1 gene (ALK1) in addition to bone morphogenetic protein receptor II gene (BMPR2) in children with pulmonary arterial hypertension. Circ J 2008, 72:127–133.
Atkinson C, Stewart S, Upton PD, et al.: Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002, 105:1672–1678.
Nakaoka T, Gonda K, Ogita T, et al.: Inhibition of rat vascular smooth muscle proliferation in vitro and in vivo by bone morphogenetic protein-2. J Clin Invest 1997, 100:2824–2832.
Long L, Crosby A, Yang X, et al.: Altered bone morphogenetic protein and transforming growth factor-beta signaling in rat models of pulmonary hypertension: potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation 2009, 119:566–576.
Toshner M, Voswinckel R, Southwood M, et al.: Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am J Respir Crit Care Med 2009, 180:780–787.
Humbert M, Morrell NW, Archer SL, et al.: Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 2004, 43(12 Suppl S):13S–24S.
Newman JH, Phillips JA 3rd, Loyd JE: Narrative review: the enigma of pulmonary arterial hypertension: new insights from genetic studies. Ann Intern Med 2008, 148:278–283.
Reynolds AM, Xia W, Holmes MD, et al.: Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2007, 292:L1182–L1192.
Zaiman AL, Podowski M, Medicherla S, et al.: Role of the TGF-beta/Alk5 signaling pathway in monocrotaline-induced pulmonary hypertension. Am J Respir Crit Care Med 2008, 177:896–905.
Morty RE, Nejman B, Kwapiszewska G, et al.: Dysregulated bone morphogenetic protein signaling in monocrotaline-induced pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol 2007, 27:1072–1078.
Sun CK, Lee FY, Sheu JJ, et al.: Early combined treatment with cilostazol and bone marrow-derived endothelial progenitor cells markedly attenuates pulmonary arterial hypertension in rats. J Pharmacol Exp Ther 2009, 330:718–726.
DeMarco VG, Habibi J, Whaley-Connell AT, et al.: Rosuvastatin ameliorates the development of pulmonary arterial hypertension in the transgenic (mRen2)27 rat. Am J Physiol Heart Circ Physiol 2009, 297:H1128–H1139.
Raoul W, Wagner-Ballon O, Saber G, et al.: Effects of bone marrow-derived cells on monocrotaline- and hypoxia-induced pulmonary hypertension in mice. Respir Res 2007, 8:8.
Zhao YD, Courtman DW, Deng Y, et al.: Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: efficacy of combined cell and eNOS gene therapy in established disease. Circ Res 2005, 96:442–450.
Newman JH, Fanburg BL, Archer SL, et al.: Pulmonary arterial hypertension: future directions: report of a National Heart, Lung and Blood Institute/Office of Rare Diseases workshop. Circulation 2004, 109:2947–2952.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bradford, C.N., Ely, D.R. & Raizada, M.K. Targeting the Vasoprotective Axis of the Renin-Angiotensin System: A Novel Strategic Approach to Pulmonary Hypertensive Therapy. Curr Hypertens Rep 12, 212–219 (2010). https://doi.org/10.1007/s11906-010-0122-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11906-010-0122-6