
Abstract
Mosquito-borne flaviviruses (MBFVs) are important cause of emerging and re-emerging human diseases nearly worldwide, transmitted by arthropod vectors (mostly aedes and culex mosquitoes), with particular reference to yellow fever virus, Japanese encephalitis virus, dengue fever virus, St. Louis encephalitis virus, Murray Valley encephalitis virus, etc. In over 100 countries, more than 2.5 billion people are at risk of infection, and approximately 20 million infections are reported annually. Through the analysis of gene sequence data of these virus populations it is possible to infer phylogenetic relationships, which in turn can yield important epidemiological information, including their demographic history. Early attempts to define the evolutionary relationships and origins of viruses in the genus flavivirus are hampered by the lack of genetic information particularly amongst the MBFVs. In this study, complete genome, translated polyprotein, structural and non-structural proteins of MBFVs have been targeted and revealed an extensive series of clades defined by their epidemiology and disease associations. The branching patterns of at the deeper nodes of the resultant trees were different from those reported in the previous study. The significance of these observations is discussed.
Similar content being viewed by others

Introduction
Mosquito-borne flaviviruses (MBFVs) are a group of viruses belonging to the genus flavivirus and the family flaviviridae causes an enormous health burden to people living in tropical and subtropical regions of the world. These diseases include dengue, yellow fever, West Nile fever, Japanese encephalitis, St. Louis encephalitis, Murray Valley encephalitis, etc. Flavivirus genus comprises approximately 70 RNA viruses, among these viruses, 36 are mosquito-borne, 16 are tick-borne and 18 are with no known vector (NKV); 22 of the 36 mosquito-borne and 13 of the 16 tick-borne flaviviruses are associated with human disease. MBFV contain dengue viruses serotypes 1–4 (DENV 1–4), yellow fever virus (YFV), West Nile virus (WNV), Japanese encephalitis virus (JEV), Murray Valley encephalitis virus (MVEV), St. Louis encephalitis (SLEV), etc. (Han et al.1999; Heinz and Mandl 1993). They are mainly transmitted by the bites of hematophagous arthropods generally female aedes and culex mosquitoes. MBFVs are widely distributed throughout Africa, the Middle East, parts of Europe, Russia, India, Indonesia, and North America (Calisher et al. 1989). The WHO estimated more than 50 million, 200,000, and 50,000, for DENV, YFV, and JEV, annual cases respectively. Severe manifestations of MBFV disease include, hemorrhaging fever (for YFV and DENV), encephalitis and neurological sequelae (for JEV, WNV, SLEV and MVEV). Extensive research has been carried out to understand these viruses and to devise ways to effectively treat the diseases caused by these MBFVs (Sampath and Padmanabhan 2009). However, no effective anti-viral treatment against flaviviruses is currently available. With that phylogeny prediction approach we are making an effort to encounter the clue for the cure from the diseases caused by MBFV group.
MBFV share a common size (40–65 nm), symmetry (icosahedral nucleocapsid) and lipid-envelop. These viruses contain a single-stranded positive-sense RNA genome, approximately 11 kb in length and appearance in the electron microscope. The genome contains a single long open reading frame (ORF) flanked by 5′- and 3′-untranslated regions. Translation of the genome generates a polyprotein that is co-translationally and post-translationally processed by the virus-encoded serine protease, NS2B/NS3, host-encoded proteases, signalase and furin, to produce the three structural proteins and seven non-structural (NS) proteins in the order C-prM/M-E -NS1-NS2A-NS2B-NS3-NS4A-NS4B-NS5 (Rice et al. 1985). The structural proteins constitute the viral particle while the nonstructural proteins are involved in viral RNA replication, virus assembly, and modulation of the host cell responses (Lindenbach et al. 2007). The E protein is a major flavivirus antigenic determinant and involved in attachment and entry of the virion to the cell. The NS protein NS3 and NS5 are the best characterized proteins, with multiple enzyme activities that are required for viral replication. NS3 has three distinct activities: serine protease together with the cofactor NS2B, required for polyprotein processing; helicase/NTPase activity, required for unwinding the double-stranded replicative form of RNA; RNA triphosphatase, required for capping nascent viral RNA (Falgout et al. 1991; Zhang et al. 1992; Arias et al.1993; Li et al. 1999; Benarroch et al. 2004). NS5 is the largest and most highly conserved flaviviral protein, with more than 75 % sequence identity across all DENV serotypes. It contains two distinct enzymatic activities, separated by an interdomain region: an S-adenosyl methyltransferase (SAM) (Grun and Brinton 1986; Chu and Westaway 1987; Tan et al. 1996; Ackermann and Padmanabhan 2001; Guyatt et al. 2001). Early attempts to define taxonomic relationships within the genus were based on antigenic cross-reactivity in neutralization, complement fixation and haemagglutination tests. Some other studies were conducted using sequences of individual genes and/or ORF to investigate the genetic relationship. The major factors that limit the quality of phylogenetic analysis with related, but widely divergent viruses are the amount of genetic information obtained for each virus, the suitability of the genomic region selected for analysis and the availability of appropriate analytical methods. In recent years, many novel MBFVs have been discovered, and this indicates larger heterogeneity among flaviviruses than previously thought and suggests that a large number of distantly related flaviviruses exist.
In the current study, to determine the phylogenetic relationships among the MBFVs with as much accuracy as possible, we undertook a comprehensive phylogenetic analysis involving complete genomes sequences, polyprotein sequences and multiple genes sequences (E protein, NS3 and NS5) reported in public database till date. These new data set provided an opportunity to extend current phylogenetic analyses and to re-examine the taxonomy of the MBFVs. At the deepest nodes of the evolutionary tree, our analysis suggests a complex relationship between viruses infecting mosquito vectors and the disease association.
Materials and methods
Sequence datasets information
The majority of the nucleotide and protein sequence data set used in study were retrieved from National Center for Biotechnology Information (NCBI) (Wheeler et al. 2007) and some of them were retrieved from the RNA virus database (http://tree.bio.ed.ac.uk/rnavirusdb/). The complete genome sequences of MBFVs were collected from viral genomes resource (NCBI) (www.ncbi.nlm.nih.gov/genomes/VIRUSES/viruses.html; Bao et al. 2004) in GenBank format (Benson et al. 2010) using RefSeq data (Pruitt et al. 2007). Several MBFVs have more than one genome isolates, so one conserved genome has been identified and used in the study. The amino acid (AA) sequences of translated polyprotein were compiled from NCBI Protein database (http://www.ncbi.nlm.nih.gov/protein) in GenPept format. The gene sequences of E, NS3 protein and NS5 protein were downloaded from viral genomes resource (NCBI) (Table 1).
Genetic characterization
Potential cleavage sites were identified according to the proteolytic processing cascade pattern for the MBFV ORF (Chambers et al. 1990). The highest cleavage potential scores obtained by SignalP-NN computer program (Chang et al. 2000) were used for determining the sites cleaved by the host cell-encoded signallase. Predicted glycosylation and cysteine residue sites were determined using the NetNGlyc (v.1.0) (http://www.cbs.dtu.dk/services/) and Protean (v. 5.03) of the LaserGene program (DNA Star), respectively. The associations of protein sequences of MBFV with other flavivirus protein were compared using the NCBI-BLAST program. The BLAST (Basic Local Alignment Search Tool), (Altschul et al. 1990) implemented via the NCBI website (www.ncbi.nlm.nih.gov/blast/) for relatedness of newly characterized sequences was evaluated against the complete Genbank database. The BlastN (Nucleotide query—Nucleotide database comparison) and BlastP (protein query—protein database comparison) in which conditional composition score adjustment having no filters of BLOSUM 62 matrix with threshold expect value 10 were used.
Sequence alignments and phylogenetic reconstruction
The alignments of nucleotide or AA sequences were generated with the help of Clustal X (1.81) program (Thompson et al. 1997) and pairwise genetic distances were estimated with the program MEGA v3.0 (Kumar et al. 2001). The phylogenetic analysis was performed using PHYLIP (phylogenetic inference program) package (version 3.57c), with the neighbour-joining (NJ) (Saitou and Nei 1987) and maximum parsimony (MP) (Swofford 2002) methods. For NJ, a distance matrix calculated from the aligned sequences by Kimura Two Parameter Formula (Kimura 1980) was used, and a weight of four for transitions versus one for transversion was selected. In MP, in order to obtain the most parsimonious tree, the heuristic algorithm was performed; and for determining the reliability of tree topology bootstrap analysis was carried out on 1,000 replicas. Bootstrap resampling technique was then used to further evaluate the reliability of the bootstrap analysis with a confidence value of 0.95 (95 %).
Results
Sequence determination and analysis
Although 36 MBFVs have reported but 22 full-genome sequence information (sequence length range 10,650–11,066 nt) are available till date. These were retrieved from NCBI and prepared for analysis. Eleven virus species out of 22 have a number of genome isolate (Supplementary Table S1). Thus total 12,298 complete genomes isolates were identified and downloaded from NCBI virus genome repository. The conserved genome for each virus species has been identified through multiple sequence alignment method using CLUSTAL-W program (Thompson et al. 1994). Full-genome of MBFVs has been produced single ORF and after translation it generates polyprotein. Twenty-two translated polyproteins were generated via ORFs, further ten other polyprotein sequences available at NCBI were also retrieved. Therefore total 32 polyprotein sequences were identified and found appropriate for the study. Antigenically important E protein is the major structural protein, plays a role in virion assembly, receptor binding and membrane fusion. Twenty-four gene sequences of E protein were generated and evaluated with the database. The NS3 protein has limited sequence information; only 12 gene sequences were available at public database. The NS5 proteins have significant sequence information. Thirty-six gene sequences of NS5 proteins were preferred and retrieved from NCBI database and comparative phylogenetic tree was generated.
Genetic characterization of polyprotein
All 12 cleavage sites for each MBFV polyprotein were identified, and all showed nearly same genome organization, with three structural and seven nonstructural proteins encoded. The results are summarized in Table 2, and the lengths of complete ORFs and deduced viral proteins are reported in Table 3. No differences in protein residues flanking the cleavage sites were found between the isolates B3 and B31. Whereas the first or second protein residues directly flanking the protein cleavage sites were mostly conserved among all MBFV, differences were found in AA residues not directly flanking the cleavage site. All sites cleaved by the viral serine protease (VirC/AnchC, NS2A/NS2B, NS2B/NS3, NS3/NS4A, NS4A/2K, and NS4B/NS5) occurred after two C-terminal basic residues, such as KR, RR, or QR. The residues flanking the sites that are cleaved by the host protease (AnchC/Pr, Pr/M, M/E, E/NS1, and 2K/NS4B) were more similar among MBFVs (Tables 2, 3).
The putative cleavage site analysis of culex-borne flavivirus and aedes-borne flavivirus were done on the basis of their clades assumed in the study. The culex-borne flavivirus group associated with 15 virus species that subdivided in the four classes was highly genetically divergent. AROAV and BSQV belong to the aroa virus clade and have same cleavage sites, where as IGUV is also the member of same clade but have difference in VirC/AnchC, AnchC/prM, AnchC/prM, M/E, E/NS1, NS3/NS4A and NS4B/NS5 cleavage sites. JEV clade comprised with seven virus species, associated to culex-borne flavivirus group were contain very much similar cleavage sites such as M/E, E/NS1, NS2A/NS2B and NS3/NS4A. NS4A/2K site have 100 % sequence similarity among all JEV clades. KOKV clade contain only one virus species i.e. KOKV, closely related with the members of AROAV clade and cleavage sites NS1/NS2A, NS3/NS4A, NS4A/2K, 2K/NS4B, NS4B/NS5 were much similar to the AROAV clade. The last clade of culex-borne flavivirus group was NTAV clade, comprised with four virus species. It has similarity in cleavage sites M/E, E/NS1, NS3/NS4A and NS4A/2K. The cleavage position NS3/NS4A and NS4A/2K have highest similarity among all culex-borne flavivirus group. The aedes-borne flavivirus group is made up of 17 exceedingly genetically similar virus species, can be separated in the three clades. The DENV clade is composed with four virus serotype 1–4 highly genetically contrary. But its potential cleavage site NS2A/NS2B, NS3/NS4A and NS4A/2K have much resemblance. KEDV, SPON and ZIKV belong to spondweni virus clade and are member of aedes-borne flavivirus group. Its M/E, NS2A/NS2B, NS3/NS4A and NS4A/2K cleavage sites encompass a lot of connection. The largest clade of MBFV is YFV clade, covers ten genetically related virus species. Its potential cleavage sites VirC/AnchC, NS2A/NS2B, NS2B/NS3, NS3/NS4A, NS4A/2K and NS4B/NS5 have nearly similar. The potential cleavage sites of whole MBFV group are confirmed that M/E, E/NS1, NS2B/NS3, NS4A/2K and 2K/NS4B having much resemblance (Table 2).
The other investigation has been done through AA residue length of proteins and found; the culex-borne flavivirus group with 3,410–3,434 AA comprises of 15 viruses and has been separated into four subgroups. In Aroa virus clade, AROAV and BSQV have similar polyprotein sequence length i.e. 3,429 AA, but IGUV have shorter sequence length (3,416 AA) in the clade. Variations have been found in the length of VirC, AnchC, NS5, NS4B and NS5 proteins. KOKV clade contain only KOKV with 3,410 AA and its size of structural and nonstructural proteins were dissimilar with other virus member of culex-borne flaviviruses group. In JEV clade ALFV, MVEV and USUV have similar polyprotein length (3,434 AA), KUNV and WNV also have the equal sequence length (3,433 AA) and JEV and SLEV have unlike sequence length i.e. 3,432 and 3,429 AA respectively. They have almost similar length of prM, M, E, NS1, NS2B, NS4A, 2K, NS4B and NS5 proteins. NTAV clade was closely related with JEV clade having four virus members. ROCV and TMUV have the same polyprotein sequence length (3,425 AA) while BAGV and ILHV encompass diverse length of polyprotein sequence i.e. 3,426 and 3,424 AA respectively. The M, E, NS2B, NS3 and NS4A proteins were identical in length. The aedes-borne flaviviruses group restrains DENV clade include all four serotypes of dengue specifically DENV1, DENV2, DENV3 and DENV4 with polyprotein length 3392, 3391, 3390 and 3387 AA respectively, their cleaved protein length were almost same except NS3 and NS4B. The SPOV clade contain KEDV (3,408 AA), SPOV (3,429 AA) and ZIKV (3,423 AA) virus members and NS1, NS2A, NS2B, NS3 and 2K proteins were nearly analogous. In YFV clade BOUV, JUGV, POTV and SABV have same residue length (3,390 AA); BANV and UGSV cover similar residue length (3,393 AA); SEPV and WESSV also have equal residue length (3,405 AA); YFV and EHV have different polyprotein lengths i.e. 3,411 and 3,410 respectively. The cleaved proteins length of YFV clade were clarify the similarity in prM, M, E, NS2B, NS4A, 2K and NS5 proteins.
Phylogenetic analysis
Construction of phylogeny using full genome and polyprotein
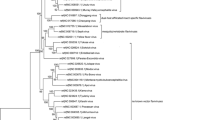
Currently, there are no comprehensive phylogenetic studies reported for entire MBFV group in systematic way. This is certainly due to the immense variability of both genomic and protein sequences within this group. In order to evaluate relatedness-by-speciation relationships among MBFV members, we constructed phylogenetic trees using the entire genomic and proteomic sequences through NJ and MP methods, evaluating node confidence values through bootstrapping using 1,000 replicates. In order to above, intact genomic sequences of MBFVs were retrieved from public database and aligned. Each NJ and MP tree was generated, and bootstrap resampling with 1,000 replicates was employed to place approximate confidence limits on individual branches (Thompson et al. 1994). The tree topologies generated from the NJ and MP methods (Fig. 1a) was correlated closely to those previously reported tree (Billoir et al. 2000; Cook and Holmes 2006; Kuno and Chang 2006; Medeiros et al. 2007; Grard et al. 2007). The unrooted phylogenetic tree was clustered into three groups and to investigation of deepest nodes assumed that the tree separated into six clades namely, AROAV, DNEV, JEV, KOKV, NTAV and YFV. Other phylogenetic tree analysis was also finished by 32 polyprotein sequences and found that the tree has been divided into four groups and deepest nodes were divided into seven clades. The evaluation of both trees produced that the SPOV clade was additional clade in the tree created by polyprotein sequences. The SPOV clade narrowly related to DENV clade and contains three virus species KEDV, SPOV and ZIKV. The full genome sequence of KEDV and ZIKV was available and tree illustrates as member of DENV clade, while the polyprotein sequence of all three virus members was available at public database and tree split into clade.

Phylogenetic tree of MBFV group computed from full genome as well as complete AA sequence using NJ method. a The phylogeny tree constructed on the bases of complete genome sequence using NJ method and tree divided into six clades. The brackets represent the virus clades and the group of virus species exposed with red color stand for the virus transmitted by culex mosquito vector with encephalitic disease; green color denotes that the virus transmitted through aedes mosquito and caused haemorrhagic disease. b The phylogenetic tree reconstructed using polyprotein protein sequences and tree segregated in seven clades. The brackets denote the virus cluster and the red color stand for the virus caused encephalitic disease and transmitted by culex mosquito vector; green color denotes the virus causes haemorrhagic disease and transmitted through aedes mosquito
An analysis and comparison of both the trees were completed on the strength of the disease association and vector responsible for transmission of MBFVs. The investigation estranged the tree into two lineages; first lineage includes the viruses associated with hemorrhagic complications and transmission by aedes species mosquitoes. The lineage has been separated to three clades; include viruses belonging to the YFV clade, DENV clade and viruses of the SPOV clade (illustrated with green color in Fig. 1). The second lineage includes a large number of viruses connected with encephalitic disease and transmitted by culex mosquitoes (show with red color in Fig. 1). The culex-borne flaviviruses have been divided into four clades namely JEV, NTAV, KOKV and AROAV clade. Thus seven clades of MBFVs, specifically, AROAV, DENV, JEV, KOKV, NTAV, SPOV, and YFV clades were recognized.
Phylogenetic analysis using E, NS3 and NS5 genes
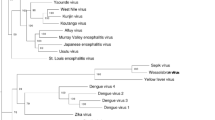
Comparative phylogenetic trees based on the gene sequences of E, NS3 and NS5 gene were produced and compared using NJ and MP methods (Fig. 2). The tree produces different branching patterns at the deepest nodes. The phylogenetic tree of E gene were generated using 24 gene sequences available at database and the tree clustered into three groups. The analysis of deepest nodes assumed that the tree has been separated into six clades (Fig. 2a). Other Phylogenetic trees were also produced using conserved gene sequence of NS3 and NS5 proteins. The NS3 gene has limited sequence information at NCBI database, only 12 gene sequences were identified, retrieved and used in tree construction (Fig. 2b). The tree has been divided in two parts and analysis of deepest node illustrated in three clades. The NS5 gene contains significant sequence information at database. Thirty six gene sequences of NS5 protein have been identified suitable for the study. The tree illustrated three main branches and the deepest node designated in seven clades (Fig. 2c). Evaluation of all three trees was completed and found almost similar tree topology but differences in branching patterns at deeper nodes (Fig. 2). The phylogenetic trees were also analyzed on the basis of vector transmission and disease association. The tree formed two distinct clusters; aedes-borne flavivirus (designated with green color in Fig. 2) and culex-borne flavivirus (signified by red color in Fig. 2). Aedes clusters of MBFVs are normally associated with haemorrhagic diseases, while culex clades are commonly associated with encephalitic diseases.

Phylogenetic analysis based on gene sequence of E protein, NS3 and NS5 protein sequences specified with (a), (b) and (c) respectively. Phylogenetic reconstruction was performed using the NJ method. The culex-borne flavivirus group is highlighted in red, the aedes-borne flavivirus group in green
Discussion
Early efforts to describe the flavivirus interrelationships and their evolutionary characteristics were based on antigenic cross reactivity in neutralization, complement fixation and haemagglutination inhibition tests (Madrid and Porterfield 1974; Calisher et al. 1989). Several other studies (Kuno et al. 1998; Gaunt et al. 2001; Cook and Holmes 2006; Billoir et al. 2000) were conducted using sequences of individual genes and/or ORF to investigate the flavivirus genetic relationship. These studies generated basically two contrasting phylogenies, NS5 gene tree and NS3/ORF tree. Classification schemes based on these criteria have proved helpful in understanding the flaviviruses, but many of the viruses have subsequently been shown to be incorrectly assigned within the schemes. Molecular sequencing and phylogenetic reconstructions have largely overcome these problems and have provided important insights into the taxonomy and dispersal of flaviviruses (Gould et al. 1997). The association of specific flaviviruses with particular arthropod vectors and vertebrate hosts has been defined precisely and a list of these characteristics for each virus is available in the International Catalogue of Arboviruses. Despite these extensive data, there have been few previous attempts to correlate molecular evolution with epidemiological and ecological features of MBFVs. The phylogenetic trees presented here have extended previous analyses of the flavivirus NS5 (Kuno et al. 1998; Billoir et al. 2000), E gene (Marin et al. 1995), full genome and NS3 phylogenetic trees (Cook and Holmes 2006). By mapping these biological characteristics onto the trees, the phylogenetic study presented in this paper demonstrates a striking series of associations between molecular phylogeny and vector responsible for transmission of virus. It was demonstrated previously (Kuno et al. 1998; Marin et al. 1995) that the flavivirus genus was monophyletic and three separate groups of viruses, namely tick-borne, mosquito-borne and NKV viruses diverge at the deepest nodes.
In present analysis we have demonstrated the most comprehensive phylogenetic study of MBFV, using complete genome, translated AA sequences, gene possessing antigenically important traits (E gene) and conserved genes (NS3 and NS5). The MBFVs are large and divergent group of viruses currently include 36 recognized species; among 36, only 22 viruses have been fully sequenced thus far. Therefore, for a better understanding of the genetic relationship among MBFVs 32 translated polyprotein were employed and analyzed. Within the viral polyproteins, proteolytic cleavage sites for the viral serine protease appeared to be highly conserved among all MBFVs studied. The prM cleavage site sequence (Arg-X-Arg/Lys-Arg) (Rice 1996) was also conserved in all genomes studied. This cleavage may be mediated by the host enzyme furin or an enzyme of similar specificity (Steiner et al. 1992; Stadler et al. 1997). The putative sites of other proteolytic cleavages, supposed to be mediated by host signalases, were less conserved, except for the M/E and 2K/NS4B cleavage site. They were only determined on the basis of sequence alignment with previously determined cleavage site sequences (Chambers et al. 1990).
Among culex-borne flavivirus cluster, AA length of prM, M, NS1, NS2B, NS4A and 2K protein sequences were found similar in AROAV and JEV clade but some variation occurred in KOKV and NTAV clade. The E protein was found conserved between entire culex-borne flavivirus and NS5 protein was most highly conserved protein among JEV clade. The AA length of M, E, NS2B and 2K were established same among aedes-borne flavivirus. The DENV and YFV clades were illustrated much resemblance in AA length of E and NS5 protein. The four serotype of DENV, belong to same clade have much similarity in AA length of structural and non structural protein except NS3 and NS4B. The AA sequence analysis of whole MBFV has indicated similarity in length of M, NS2B, and 2K proteins.
Considering the phylogenetic relationships, through the modes of vector transmission and disease relationship of MBFV, we propose that the mosquito-borne viruses could be divided into two epidemiologically distinct vector groups, those that were primarily isolated from aedes species and those that were primarily isolated from culex species. The MBFVs that were primarily isolated from aedes species, causes haemorrhagic disease formed three paraphyletic clade, containing YFV, SPOV and DENV, hereafter denoted as the aedes-borne flavivirus group. Other viruses in mosquito-borne group, i.e. JEV, NTAV, AROAV and KOKV, have been primarily associated with culex species causes encephalitic disease, hereafter denoted as the culex-borne flavivirus group.
According to the taxonomic proposal through the comparison of phylogenetic tree generated by full genome, polyprotein sequences and multi gene (E, NS3 and NS5) sequences, the branching pattern suggested that viruses transmitted by culex spp. mosquitoes evolved from an ancestral lineage associated with aedes spp. mosquitoes, as was previously suggested from the NS5 nucleotide sequence data (Gould et al. 2001, 2003). The complete genome phylogeny also suggests two possible taxonomic reassessments. ZIKV, KEDV and SPOV are currently recognized as a member of the SPOV clade. Both KEDV and SPOV viruses circulate in Africa but ZIKV was isolated in Asia and Oceania. They are transmitted by aedes spp. mosquitoes and can induce human epidemics. The prior studies collectively indicated that KEDV was close to DENV and currently a member of the DENV lineages, reported in phylogenies based on the NS5 gene (Kuno et al. 1998; Gaunt et al. 2001). On the other hand, our phylogeny inferred from the complete AA and multi gene (E and NS5) data suggested that KEDV is strictly associated with SPOV clade, although this was not robustly supported in the tree produced by full genome sequence (Fig. 1a), and indeed, in the polyprotein, E and NS5 gene NJ phylogenetic tree (Figs. 1b, 2a, c correspondingly) of KEDV appeared to be separated from both the DENV and SPOV clade. The phylogeny suggested that, SPOV clade is clearly related to culex borne flaviviruses except the tree created by full genome, KEDV and ZIKV is similar to viruses of both the culex group and the DENV group, again showing that its phylogenetic position is ambiguous (Figs. 1, 2). The position of dengue virus serotype in DENV clade is same in tree produced by full genome, polyprotein and NS5 sequence data, however different with the tree produced via E and NS3 gene. The phylogenetic relationships inferred here for the other members of the aedes-borne flavivirus group are same with the current taxonomic position.
The JEV clade contain predominantly neurotropic viruses belong to culex-borne flavivirus having more than 50 % species in the cluster. Several other members of culex-borne flavivirus are NTAV, AROAV and KOKV clade. Although both KOKV and AROAV clade are neurotropic and together share a sub-cluster in the phylogenetic tree, unlike the members of the neurotropic JEV clade transmitted by culex mosquitoes, both virus clade have closed association with DENV clade which belongs to aedes group. NTAV clade is narrowly linked with JEV clade but phylogeny through NS5 gene indicated that SPOV and KOKV clade are strictly associated with NTAV clade. This may be because of the limited sequence information is available in public database and therefore it is possible that KEDV, SPOV and NTAV clade belongs to a distinct group of viruses for which other members remain to be discovered.
The subsequent major correlation was between the type of disease produced and the mosquito clade in which each virus appeared. In general, severe infections caused by some aedes species viruses result in haemorrhagic disease, whereas many culex species viruses cause encephalitic disease. However, exceptions to this generalization have been reported for several MBFVs. The KOUV and SABV have been isolated more often from ticks and sandflies, respectively, and which are not known to be neurotropic. In contrast with the MBFVs, different viruses in the tick-borne virus groups produce encephalitic disease, but OHF and KFD viruses may also produce haemorrhagic disease in humans. Until the precise basis of flavivirus pathogenicity has been defined at the molecular level, it is not possible to understand why these different disease associations can be seen in the phylogenetic tree.
During the past few years our knowledge of the spectrum of flaviviruses has widened as new species in the genus flavivirus have been isolated and characterized. These new findings may be helpful in genome characterization and determination of the exact phylogenetic and taxonomic relationships of MBFVs. Such data will be essential for achieving the ultimate goals of designing better molecular probes and primers for improved surveillance and diagnosis, determination of the neurovirulence markers at a molecular level, and development of attenuated vaccine and antiviral drugs.
References
Ackermann M, Padmanabhan R (2001) De novo synthesis of RNA by the dengue virus RNA-dependent RNA polymerase exhibits temperature dependence at the initiation but not elongation phase. J Biol Chem 276(43):39926–39937
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Arias CF, Preugschat F, Strauss JH (1993) Dengue2 virus NS2B and NS3 form a stable complex that can cleave NS3 within the helicase domain. Virology 193(2):888–899
Bao Y, Federhen S, Leipe D, Pham V, Resenchuk S, Rozanov M, Tatusov R, Tatusova T (2004) National center for biotechnology information viral genomes project. J Virol 78(14):7291–7298
Benarroch D, Selisko B, Locatelli GA, Maga G, Romette JL, Canard B (2004) The RNA helicase, nucleotide 5′-triphosphatase, and RNA 5′-triphosphatase activities of Dengue virus protein NS3 are Mg2+-dependent and require a functional Walker B motif in the helicase catalytic core. Virology 328(2):208–218
Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW (2010) GenBank. Nucleic Acids Res 38:D46–D51
Billoir F, de Chesse R, Tolou H, de Micco P, Gould EA, de Lamballerie X (2000) Phylogeny of the genus Flavivirus using complete coding sequences of arthropod-borne viruses and viruses with no known vector. J Gen Virol 81:781–790
Calisher CH, Karabatsos N, Dalrymple JM, Shope RE, Porterfield JS, Westaway EG, Brandt WE (1989) Antigenic relationships between flaviviruses as determined by cross neutralisation tests with polyclonal antisera. J Gen Virol 70:37–43
Chambers TJ, Hahn CS, Galler R, Rice CM (1990) Flavivirus genome organization, expression, and replication. Annu Rev Microbiol 44:649–688
Chang GJJ, Hunt AR, Davis B (2000) A single intramuscular injection of recombinant plasmid DNA induces protective immunity and prevents Japanese encephalitis in mice. J Virol 74:4244–4252
Chu PW, Westaway EG (1987) Characterization of Kunjin virus RNA-dependent RNA polymerase: reinitiation of synthesis in vitro. Virology 157(2):330–337
Cook S, Holmes EC (2006) A multigene analysis of the phylogenetic relationships among the flaviviruses (Family: Flaviviridae) and the evolution of vector transmission. Arch Virol 151:309–325
Falgout B, Pethel M, Zhang YM, Lai CJ (1991) Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J Virol 65(5):2467–2475
Federhen S (2012) The NCBI Taxonomy database. Nucleic Acids Res 40(D1):D136–D143
Gaunt MW, Sall AA, de Lamballerie X, Falconar AK, Dzhivanian TI, Gould EA (2001) Phylogenetic relationships of flaviviruses correlate with their epidemiology, disease association and biogeography. J Gen Virol 82:1867–1876
Gould EA, Zanotto PMA, Holmes EC (1997) The genetic evolution of the flaviviruses. In: Saluzzo JF, Dodet B (eds) Factors in the emergence of arboviruses diseases. Elsevier, Paris, pp 51–63
Gould EA, de Lamballerie X, Zanotto PM, Holmes EC (2001) Evolution, epidemiology, and dispersal of flaviviruses revealed by molecular phylogenies. Adv Virus Res 57:71–103
Gould EA, de Lamballerie X, Zanotto PM, Holmes EC (2003) Origins, evolution, and vector/host coadaptations within the genus Flavivirus. Adv Virus Res 59:277–314
Grard G, Moureau G, Charrel RN, Lemasson JJ, Gonzalez JP, Gallian P, Gritsun TS, Holmes EC, Gould EA, de Lamballerie X (2007) Genetic characterization of tick-borne flaviviruses: new insights into evolution, pathogenetic determinants and taxonomy. Virology 361:80–92
Grun JB, Brinton MA (1986) Characterization of West Nile virus RNA-dependent RNA polymerase and cellular terminal adenylyl and uridylyl transferases in cell-free extracts. J Virol 60(3):1113–1124
Guyatt KJ, Westaway EG, Khromykh AA (2001) Expression and purification of enzymatically active recombinant RNA-dependent RNA polymerase (NS5) of the flavivirus Kunjin. J Virol Methods 92(1):37–44
Han LL, Popovici F, Alexander JJ, Laurentia V, Tengelsen LA, Cernescu C, Gary JH, Ion NN, Campbell GL, Tsai TF (1999) Risk factors for West Nile virus infection and meningoencephalitis, Romania. J Infect Dis 179:230–233
Heinz FX, Mandl CW (1993) The molecular biology of tick-borne encephalitis virus. Acta Pathol Microbiol Immunol Scand 101:735–745
Kimura M (1980) A simple method for estimating evolutionary rate of base substitution through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Kumar S, Tamura K, Jakobsen IB, Nei M (2001) MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17(12):1244–1245
Kuno G, Chang GJ (2006) Characterization of Sepik and Entebbe bat viruses closely related to yellow fever virus. Am J Trop Med Hyg 75:1165–1170
Kuno G, Chang GJJ, Tsuchiya KR, Karabatsos N, Cropp SB (1998) Phylogeny of genus Flavivirus. J Virol 72:73–83
Li H, Clum S, You S, Ebner KE, Padmanabhan R (1999) The serine protease and RNA-stimulated nucleoside triphosphatase and RNA helicase functional domains of dengue virus type 2 NS3 converge within a region of 20 amino acids. J Virol 73(4):3108–3116
Lindenbach BD, Thiel HJ, Rice CM (2007) Flaviviridae: the viruses and their replication. Lippincott William & Wilkins, Philadelphia
Madrid AT, Porterfield JS (1974) The flaviviruses (group B arboviruses): a cross-neutralisation study. J Gen Virol 23:91–96
Marin MS, Zanotto PM, Gritsun T, Gould EA (1995) Phylogeny of TYU, SRE, and CFA virus: different evolutionary rates in the genus Flavivirus. Virology 206:1133–1139
Medeiros DB, Nunes MR, Vasconcelos PF, Chang GJ, Kuno G (2007) Complete genome characterization of Rocio virus (Flavivirus: Flaviviridae), a Brazilian flavivirus isolated from a fatal case of encephalitis during an epidemic in Sao Paulo state. J Gen Virol 88:2237–2246
Pruitt KD, Tatusova T, Maglott DR (2007) NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35:D61–D65
Rice CM (1996) Flaviviridae: the viruses and their replication. In: Fields BN, Knipe DM, Howley PM (eds) Fields virology, 3rd edn. Lippincott-Raven, Philadelphia, pp 931–959
Rice CM, Lenches EM, Eddy SR, Shin SJ, Sheets RL, Strauss JH (1985) Nucleotide sequence of yellow fever virus: implications for flavivirus gene expression and evolution. Science 229(4715):726–733
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4(4):406–425
Sampath A, Padmanabhan R (2009) Molecular targets for flavivirus drug discovery. Antiviral Res 81(1):6–15
Stadler K, Allison SL, Schalich J, Heinz FX (1997) Proteolytic activation of tick-borne encephalitis virus by furin. J Virol 71:8475–8481
Steiner DF, Smeekens SP, Ohagie S, Chan SJ (1992) The new enzymology of precursor processing endoproteases. J Biol Chem 267:23435–23438
Swofford DL (2002) PAUP*: phylogenetic analysis using parsimony (* and other methods), 4.0b10 edition. Sinauer Associates, Sunderland
Tan BH, Fu J, Sugrue RJ, Yap EH, Chan YC, Tan YH (1996) Recombinant dengue type 1 virus NS5 protein expressed in Escherichia coli exhibits RNA-dependent RNA polymerase activity. Virology 216(2):317–325
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22(22):4673–4680
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25(24):4876–4882
Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Edgar R, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Khovayko O, Landsman D, Lipman DJ, Madden TL, Maglott DR, Miller V, Ostell J, Pruitt KD, Schuler GD, Shumway M, Sequeira E, Sherry ST, Sirotkin K, Souvorov A, Starchenko G, Tatusov RL, Tatusova TA, Wagner L, Yaschenko E (2007) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 36:D13–D21
Zhang L, Mohan PM, Padmanabhan R (1992) Processing and localization of Dengue virus type 2 polyprotein precursor NS3-NS4A-NS4B-NS5. J Virol 66(12):7549–7554
Acknowledgments
The authors are thankful to King George’s Medical University, Lucknow and Biotech Park, Lucknow for providing workspace and also thanks to Indian Council of Medical Research, New Delhi for providing financial support via grant for Senior research fellowship (IRIS ID: 2010-04490).
Conflict of interest
Authors declare no conflict of interest on the contents of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Gupta, S.K., Singh, S., Nischal, A. et al. Molecular-based identification and phylogeny of genomic and proteomic sequences of mosquito-borne flavivirus. Genes Genom 36, 31–43 (2014). https://doi.org/10.1007/s13258-013-0137-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-013-0137-x