
Abstract
Mechanoresponses in mesenchymal stem cells (MSCs) guide both differentiation and function. In this review, we focus on advances in our understanding of how the cytoplasmic cytoskeleton, nuclear envelope, and nucleoskeleton, which are connected via linker of nucleoskeleton and cytoskeleton (LINC) complexes, are emerging as an integrated dynamic signaling platform to regulate MSC mechanobiology. This dynamic interconnectivity affects mechanical signaling and transfer of signals into the nucleus. In this way, nuclear and LINC-mediated cytoskeletal connectivity plays a critical role in maintaining mechanical signaling that affects MSC fate by serving as both mechanosensory and mechanoresponsive structures. We review disease- and age-related compromises of LINC complexes and nucleoskeleton that contribute to the etiology of musculoskeletal diseases. Finally, we invite the idea that acquired dysfunctions of LINC might be a contributing factor to conditions such as aging, microgravity, and osteoporosis and discuss potential mechanical strategies to modulate LINC connectivity to combat these conditions.
Similar content being viewed by others

Introduction
To thrive, living organisms must sense, respond, and ultimately adapt to their physical environments at the cellular, tissue, organ, and organismal levels. Adaptations are initiated at the level of the cell, where mechanosensory complexes have evolved to allow translation of mechanical signals into biologically relevant responses. In the last decade, considerable progress has been made in identifying the many mechanisms by which cells sense and respond to both static and dynamic mechanical cues by initiating signaling events that result in differential gene expression and concurrent adaptations in cytoskeletal architecture [1]. Such mechanically driven changes in cellular structure not only ensure that forces are effectively transmitted into the nucleus, but also dynamically tune nucleoskeletal architecture and associated gene expression, thus physically influencing the biological response [2]. In the case of pluripotent cells, these processes direct subsequent differentiation accordingly. For instance, the mesenchymal stem cells residing in bone marrow respond to external loading during physical activity with allocation to osteoblast lineage and subsequent bone formation, enabling an appropriate adaptive response at the tissue level to functional challenges [3–5]. When the mechanosensing capacity of these progenitor cells is compromised, mechanical cues fail to elicit appropriate cell fate decisions, ultimately contributing to the etiology of diseases such as osteoporosis.
Focal adhesions, the sensory elements that bridge the cell’s plasma membrane to the extracellular matrix, have long been accepted to play a critical role in sensing the mechanical cues of its external milieu and regulating signaling pathway responses [6]. More recently, the nuclear envelope, which establishes a barrier separating nuclear and cytoplasmic compartments, has also been recognized as a mechanosensory element that regulates both biochemical and physical coupling of the nucleus with the extra-nuclear cell cytoskeleton [7], and thus direct coupling to the cell membrane and ECM. The linker of nucleoskeleton and cytoskeleton (LINC) complexes [8] that bridge between the nucleus and the surrounding cytoskeleton serve to mechanically connect the inner nucleoskeletal lamin A/C network with the cytoplasmic cytoskeleton and are beginning to be recognized to have significant regulatory effects on cell differentiation and function [9, 10]. Recent advances in our understanding of how nuclear envelope and nucleoskeleton associated elements contribute to both health and disease [11, 12] make it clear that mesenchymal tissues in particular require a functional nuclear envelope to adequately respond to mechanical challenges.
In this review, we will focus on how mechanical force generates signals at the interface of the nucleus and the cytoplasm that define lineage output from mesenchymal stem cells (MSC). In particular, we will concentrate on recent understanding of how the cytoplasmic cytoskeleton, nuclear envelope, and nucleoskeleton, which are connected via LINC complexes, are emerging as a combined dynamic signaling platform to enable MSC mechanobiology. As importantly, decline in the LINC complex and associated nucleoskeleton, as occurs with aging, disuse, or disease, suppresses the adaptive capacity of the cell and may contribute to the etiology of diseases such as osteopenia, sarcopenia, progeria, and obesity. We believe that mechanically improving connectivity in the cell may be a strategy to improve MSC functionality.
LINC Complex Structure
LINC complexes provide the physical hardwiring of the cell nucleus to the outside world. These complexes allow the nucleus to perceive the extranuclear mechanical environment by tying the nucleus to the cytoplasmic structure through interactions with actin, microtubule, and intermediate filament networks [13]. A LINC complex is made of multiple components. Giant nesprin proteins 1 and 2 provide the cytoplasmic face of the LINC complex. N-termini of giant nesprins share a CH domain identical to that found in α-actinin, binding actin with high affinity [14, 15]. Further, the CH domain of nesprin promotes actin polymerization in vitro [15], potentially increasing cytoplasmic structure. Through interactions with kinesin and dyenin, nesprin on the cytoplasmic face of the nucleus also interacts with microtubules [16, 17]. Spectrin repeat (SR) domains connect nesprin’s N and C terminal domains: The nesprin-1 giant possesses 72 SR domains while nesprin-2 giant has 56 SR domains [18]. Although these large SR regions are thought to play a role in mechanical load transmission [19], little is known about how they do so. Computational studies suggest that the highly conserved SR domains [18] present putative binding sites for protein–protein interactions [19]. Interestingly, almost 30 % of all SRs in giant nesprins display buried residues which point to possible mechanically functional regions for dynamic protein–protein interactions. The C-termini of the nesprin proteins pierce through the outer nuclear membrane via a transmembrane domain to end in a highly conserved Klarsicht, ANC-1, Syne Homology (KASH) domain [20]. Overexpression of nesprin-mini, a nesprin designed to lack the large SR rod domain, decreases nuclear size [21]. This suggests that as opposed to the popular depiction in the field that shows nesprin filaments protruding out of the nuclear surface, giant nesprin might instead form a filamentous network that wraps around the nucleus and restricts nuclear size. Along with giant nesprins, there are multiple smaller nesprin-1/2 isoforms that share KASH and tail domains but lack CH and large SR regions [22]. These smaller α and β isoforms are found in both cytoplasm and nucleus [23]. Nesprin-3 is structurally distinct from giant isoforms but provides nuclear connection with intermediate filaments through its interaction with plectin [13].
The KASH domain of giant nesprin binds to the Sun (Sad1p, UNC-84) proteins that span the nuclear leaflet and emerge within the nucleus where they bind to the inner lamin A/C network. In mammalian cells, two isoforms, Sun1 [24] and Sun2 [25], are expressed whose protein length is predicted to be similar to the distance between outer and inner nuclear membranes [26, 27]. In mechanically specialized cells, shortened forms of Sun proteins decrease the spacing between inner and outer nuclear membranes, while complete deletion of Sun proteins leads to increase in the envelope thickness [28]. Structural analysis of Sun proteins reveal that Sun1 and Sun2 form a strong trimeric structure which then binds to three KASH peptides to effectively interconnect multiple proteins making up the LINC complexes [29]. Deletion of either Sun1 or Sun2 alone does not disrupt LINC connections [8] suggesting that these two isoforms share a similar function. But, although Sun1 and Sun2 share a very similar structure with perhaps overlapping function [30], anchorage of Sun2 to the nuclear envelope is mediated by lamin A/C while Sun1 anchors in the absence of lamin A/C [31]. Functionally, Sun1 appears to be critical in meiosis, and Sun2 may be more heavily recruited in the formation of higher-order structures such as the LINC complex [32]. Further, Sun1, but not Sun2, is found to be directly associated with nuclear pore complexes (NPC) [33].
Within the nuclear envelope and inside the nucleus, emerin is a functionally important binding partner of LINC [34]. Although emerin has not been shown to be required for the structural integrity of LINC, its mutations lead to a range of musculoskeletal syndromes that are reminiscent of LINC envelopathies [35, 36]; this implicates emerin in the regulation of LINC function [37]. Emerin is an actin capping protein that binds to the pointed ends of actin fibers and can cause accelerated actin polymerization [38]. Its nuclear localization and minimal effects on cytoplasmic actin support the idea that emerin’s role is in nuclear actin assembly [39]. Emerin also complexes with α-catenin, β-catenin, and LINC complexes and plays a role in the export of nuclear β-catenin as the deletion of emerin increases β-catenin nuclear accumulation [40–43]. Further, emerin is phosphorylated by Src kinases upon mechanical stimulation, which leads to increased nuclear stiffness in a lamin A/C-dependent manner [44]. This suggests that emerin, as part of its association with LINC, is a critical component in translating mechanical information to the nucleus.
Finally, nuclear structure depends on a lamin nucleoskeleton which receives transmitted force via LINC connections. Lamin is an intermediate filament that forms a continuous meshwork just inside the inner nuclear membrane [45]. Functionally, lamins are classified as lamin B and lamin A/C. Lamin B, encoded by Lmnb gene [46], can be found in virtually every cell and is constitutively expressed [45]. Lamin A and lamin C isoforms are different splice variants of the Lmna gene [47]. As opposed to lamin B, lamin A/C is found at low levels in undifferentiated pluripotent cells and is expressed as differentiation ensues [48]. Lamin A/C provides the majority of the measured mechanical strength of the nuclear membrane and behaves like a viscoelastic network [49–51]. As opposed to chromatin, which is thought to provide the compressive resistance of the nucleus during large deformations [52], lamin A/C dominates the mechanical behavior of nuclear mechanics under small deformations. Interestingly, lamin A/C protein levels have been shown to be correlated with tissue stiffness [53] suggesting that as cells become more mechanically competent—or tougher—they may require more robust lamin A/C networks to function in a stiff mechanical environment. Indeed, lamin deficiency or dysfunction leaves cells more susceptible to mechanical damage, as in Hutchison–Guilford progeria where tissues experiencing mechanical stress “age” more rapidly, i.e., cardiac muscle, skin, and skeleton [54], preventing them from responding and adapting to mechanical challenges adequately. Lamin A/C also serves as an intranuclear anchoring network for many proteins including Suns and emerin [55]. Partial or complete loss of lamin A/C has been shown to limit LINC-mediated nuclear-cytoskeletal connectivity [37] ultimately contributing to the etiology of many musculoskeletal conditions by limiting the mechanical coupling of nucleus to the outside world.
The Nucleus as a Mechanically Regulated Organelle
The nuclear envelope transmits mechanical force from the extracellular matrix inward to the nucleus [56], and these mechanical signals alter the shape and location of nuclear lamin [57]. As such, the structural relationship of lamin A/C with nuclear heterochromatin [58] affects the accessibility and action of transcriptional regulators [10]. While transcriptionally active euchromatin is found at the center of the nucleus, transcriptionally silent chromatin sites attach to lamin A/C at the periphery of the nucleus [10]. Although the mechanisms by which lamin A/C might regulate chromatin function or activity are not clear, it is interesting to speculate that beyond mechanical competency, a more robust lamin A/C network in mechanically specialized cells may regulate gene activity by controlling gene accessibility to transcription factors, or even direct access to heterochromatin. Interestingly, deletion of nesprin-2 alters perinuclear localization of tri-methylated histone H3K9Me3 in keratinocytes which results in delayed wound healing response, presumably due to altering gene access [59]. The loss of LINC complex and lamin A/C in cancer cells is supportive of the idea that lamin A/C and LINC complex play important roles in chromatin dynamics to control cell fate, such that the loss of nuclear structure leads to gene unsilencing and aberrant proliferation [60•]. One of the most interesting examples highlighting the importance of mechanical cues in nuclear chromatin dynamics is seen in embryonic stem cells that present an open chromatin state [61] and express very low levels of lamin A/C [62]. During the transition phase of embryonic stem cells from undifferentiated to a pre-differentiation stage, the nucleus becomes auxetic, an intrinsic mechanical behavior described by a negative Poisson’s ratio [63] (when compressed longitudinally, material cross section becomes smaller in transverse axis) effectively increasing compressive resistance [64]. This suggests that during the cellular re-programming phase, the auxetic behavior of the nucleus mechanically shield intranuclear compartment to avoid confounding mechanical effects.
Maintenance of nuclear shape and lamin A/C polarization by the compressive forces generated by the perinuclear actin cytoskeleton [65, 66•] suggests another level of cytomechanical control on heterochromatin: specifically, physical changes in the nucleoskeleton may alter the nonrandom organization of chromosomes [67••]. The nucleoskeleton can adapt to mechanical qualities of the surrounding matrix as well as dynamic forces. Fluid shear stress shown to expose the Cys522 site in lamin A/C tail Ig domain and phosphorylation of Ser/Thr residues here appear to be inversely correlated with lamin A/C stability [53]. Increasing matrix stiffness suppresses the Ser/Thr phosphorylation and increases lamin A/C levels [68]. As noted above, in isolated nuclei, forces applied through nesprin-bound magnetic beads directly induce emerin phosphorylation to control nuclear stiffness in a lamin A/C-dependent manner [44]. Considering that lamin A/C- and LINC-deficient cells appear to be more susceptible to mechanical damage [53, 54] and higher lamin A/C levels are found in mechanically competent cells [53] suggests that lamin A/C—and by extension LINC complexes—may play a role in shielding chromatin dynamics from excessive force to maintain normal transcriptional activity. Highlighting the importance of lamin A/C and LINC complexes in transferring mechanical forces into the nucleus, magnetic bead-induced forces applied through LINC connections control the displacements of coilin and SMN proteins in nuclear Cajal bodies in a lamin A/C-dependent manner [69].
New studies suggest that the nucleus is mechanically coupled with the cytoplasmic cytoskeleton in a dynamic manner and can respond to mechanical challenges within minutes [70]. Force applied onto the nuclear surface by an actin stress fiber appears to increase the connectivity strength between the nucleus and the actin fiber [71], but the timescale in which this coupling occurs remains unclear. Thus, as opposed to a passive binding action, the nuclear envelope might act as a mechanosensory center to actively control these dynamic events. Recent evidence indicates that in addition to CH domain-mediated passive binding, nesprin-2G interacts with formin homology 2 domain containing 1 (FHOD1) through its central spectrin repeat domain to increase the coupling strength between LINC and F-actin [72]. This suggests that, similar to focal adhesions at the plasma membrane, nesprin might act as a backbone for mechanoresponsive signaling hubs on the nuclear envelope. Interestingly, it has been demonstrated in multiple cell types that F-actin bundling on the nuclear surface not only creates indentations nuclear surface but that LINC complexes and lamin A/C are recruited to such high-stress regions [73••]. Torsin A is another nuclear envelope protein that belongs to AAA+ family (ATPases associated with various cellular activities) that utilizes ATP to unfold other proteins and thus implicated in playing a role in LINC assembly/disassembly [74, 75]. Using models of dystonia, an autosomal neurological disorder, Torsin A has been shown to interact with Sun1 [76] and nesprin3α [77]. Given the functional interactions between lamina-associated polypeptide 1 (LAP-1) and Torsin A [78], as well as emerin [79], it would be interesting to hypothesize that mechanically induced emerin phosphorylation [44] might play a role in recruiting Torsin A to high-stress regions thus building a LINC complex mechanosensory hub at the nuclear envelope.
These findings demonstrate that the nucleus is not only a destination for signaling pathways, but that, through LINC-mediated mechanical integration of nucleus into the cell architecture, the nucleus is a mechanosensory and mechanoresponsive organelle (Fig. 1). In this regard, the nuclear envelope including the nucleoskeleton should be considered as integral to tissue level mechanical adaptations.

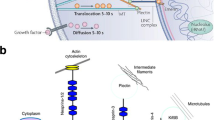
Nuclear envelope is a mechanically integrated signaling platform. Schematic of nuclear envelope including nucleoskeleton and their binding partners that potentially play a role in MSC fate selection as well as facilitating the mechanical coupling between cytoplasmic and nuclear cytoskeletons. LINC complexes composed of sun trimers and giant nesprin mechanically couple the actin cytoskeleton. For simplicity, giant nesprin isoforms were indicated as nesprin and sun trimers were drawn as monomers. Mechanical force activates RhoA-mediated cytoskeletal remodeling, potentially increasing the LINC coupling with actin. In this regard, Torsin A implicated to facilitate the LINC assembly at the nuclear envelope. Mechanical coupling of actin and LINC involves a cytoplasmic formin FHOD1 that couples the nesprin and actin at multiple points for a more robust binding. Nesprin, emerin, and β-catenin form complexes at the nuclear envelope potentially retaining β-catenin on the nuclear surface. Sun1 directly binds to nuclear pore complexes, and in this way, mechanical forces applied on LINC complex or mechanically induced recruitment of LINC to nuclear envelope may play a role in β-catenin signaling
LINC Integration of Nucleus to Cytoskeleton Regulates Cell Mechanosensitivity
Cells connect to their extracellular matrix via transmembrane integrins [80]. At the cytoplasmic side of the cell membrane, integrin attachment sites form large protein complexes called focal adhesions that serve as signaling platforms to inform cellular functions about their mechanical environment and provide anchoring points for cell cytoskeleton [81]. The cytoskeleton is the structural element within the cell that both transmits and generates force—and as such readily responds to mechanical challenges by reinforcing its structure through formation of new stress fibers [82]. Force generation in stress fibers is largely controlled by RhoA, a small GTPase [83], that increases the cell tension through its effector protein Rock, activating myosin light chain kinase (MLCK). MLCK in turn activates the dimerized motor protein myosin II [84]. We and others have shown that mechanical challenges increase the number of focal adhesions and associated cytoskeletal structure to adapt to mechanical challenges through stimulation of RhoA [85, 86]. It is interesting to note that these mechanical adaptations observed in vitro are transient in nature. For example, substrate strain, as an in vitro analog of strenuous exercise, suppresses the adipogenic pathways in MSCs, signaling events in response to substrate strain transiently increases the number of focal adhesions but a second application of substrate strain within 1–3-h results in amplification the mechanical response leading to more efficient suppression of MSC adipogenesis [87]. For further reading, there are excellent reviews that discuss the role of focal adhesions and other mechanosensory organelles—including cell cytoskeleton—in cell function and fate [88–91].
The nucleus is the biggest central element in almost all cell types, and any external signal—whether mechanical or biochemical—must ultimately reach the nucleus to influence gene expression. Mechanical signals are transmitted to the nucleus through the cytoskeleton by virtue of its connection via LINC complexes. When the cell substrate is subject to mechanical strain, for example, the nucleus—will also be deformed through LINC-mediated connections to the actin cytoskeleton [92]. Moreover, disrupting LINC function by overexpressing dominant negative forms of nesprin KASH or Sun luminal domains interrupts this mechanical connection and prevents strain-induced nuclear deformations [56]. Interestingly, depletion of nesprin-1 in endothelial cells increases the number of focal adhesions in cells suggesting a compensatory mechanism to maintain cellular structure when LINC complexes are disrupted [93].
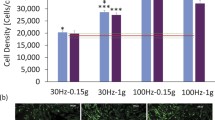
Structural integration of the nucleus into the cell architecture enabled by LINC connectivity plays a role in cell mechanosensitivity and mechanoresponse. Mechanosensitivity of MSC is reduced in aging such that these progenitors increase output of adipocytes and decrease that of osteoblasts [94, 95]; this dysfunctional MSC output is also observed in mice with lamin A/C mutations [96]. This suggests that allocation of MSCs into adipogenic lineage may be accelerated by deterioration of the LINC complex resulting in a loss of mechanosensitivity. Interestingly, Sun1- or nesprin4-deficient mice, where the nucleus is not adequately coupled to the cytoplasmic cytoskeleton, fail to maintain the basal localization of nucleus in sensory epithelial cells. This results in a gradual hearing impairments as the mice mature [97]. This suggests that mechanical vibrations, such as those sensed as sound, are received at the level of LINC. Most interestingly, we and others have shown that cells are responsive to vibration: Indeed, application of high-frequency, low-intensity vibrations (LIVs) mimics aspects of exercise to improve musculoskeletal function [98, 99] to decrease adipose encroachment in the bone marrow and to augment MSC osteogenesis [100] and improve gap junctional communication within bone cells [101]. To understand how cells sense these small mechanical signals, we recently showed that the LINC complex is critical to sensing of the high-frequency low-intensity vibrations that affect MSC lineage decisions [98, 100]. Low-intensity vibration activates focal adhesion kinase (FAK)-mediated RhoA activation and subsequent cytoskeletal remodeling in MSCs [67••]. Disabling LINC function via siRNA treatment against Sun1 and Sun2 proteins, or by expressing a dominant negative form of nesprin KASH domain, inhibited vibration-induced FAK and Akt phosphorylations pointing toward a critical role of LINC connectivity as a mechanosensor in cells. Importantly, similar FAK and Akt signaling in response to substrate strain known to arise at focal adhesions [85] was unaffected by LINC deficiency [67••]. Specifically, while the FAK co-regulator Fyn was essential for strain-induced Akt activation [86], Fyn depletion did not prevent vibration-induced Akt activation [67••]. Further, strain-induced cell response was also found to be intact in LINC-deficient fibroblast [56], but LINC-deficient cardiomyocytes showed a decreased mechanical response [102]. These findings point toward the cell’s ability to differentiate between distinct types of mechanical signals as achieved though different structures.
In addition to the transient adaptations of the cytoplasmic cytoskeleton, it is possible that the dynamic adaptations in the nuclear envelope and nucleoskeleton are responsible for regulating, and even accentuating, responses to mechanical signals. Despite the convergence of many musculoskeletal conditions on the mutations of nuclear envelope, how the nuclear envelope and nucleoskeleton are mechanically maintained is poorly understood. One interesting hypotheses is that, analogous to the positive and negative regulation of cell cytoskeleton by loading and unloading, respectively [94], LINC complexes and lamin A/C could also be mechanically modulated. The ability of LINC complexes and lamin A/C to remodel in response to changing actin stress fibers [103] suggests that mechanical influences may cause gain or loss of function in nucleoskeleton. In support of this concept, low-intensity vibration increases F-actin remodeling around the perinuclear area [67••], potentially affecting mechanical connectivity throughout the cell. Considering that nuclear envelope can serve as an F-actin nucleation surface [104], these findings suggest that mechanical therapies like low-intensity vibration may be enabled by first stimulating the formation of a more robust coupling between cytoplasmic and nuclear cytoskeletons, and thus making the cell more sensitive to mechanical or biochemical signals in general. Not surprisingly, application of low-intensity vibration not only amplifies the response to other mechanical [67••] or biochemical [105] factors like substrate strain or BMP4, but repeated exposure to low-intensity vibration is more effective than a single bout in suppressing MSC adipogenesis [106]. Generalizing to physical responses at organism level, short but repeated exercise bouts may be more effective in guiding some outcomes than a single longer-duration exercise session [107].
Role of LINC-Nuclear Connections in Bone/Osteoblast Phenotype
Bone provides protection and mechanical support to the whole body and is continuously eroded and rebuilt by a remodeling process where osteoblasts form new bone and osteoclasts resorb established bone. Although many factors such as diet, hormone levels, and age can tip this balance toward anabolic or catabolic skeletal outcomes, mechanical signals remain a singular factor in enhancing bone structure [108], with a dynamic mechanical environment recognized as essential for forming and maintaining healthy bone [109]. In addition to signaling the resident osteoblast population to stimulate matrix formation, anabolic outcomes in response to mechanical loading also depend on recruitment of bone marrow MSC into an osteogenic lineage. For example, introducing LIV into the mechanical environment has been shown to improve the fracture repair [110], where stem cells are recruited to the callus to be re-integrated into bone tissue [111] through a process mirroring the skeletal development in a smaller scale [112]. When physical loading is absent, MSCs have a tendency to enter adipogenic lineage [113], a phenotype that predominates in paraplegic, sedentary, and elderly individuals [114], achieved in a reciprocal relationship to a declining musculoskeletal system.
In bone marrow, MSCs position themselves in close proximity to bone surfaces and are continuously subjected to mechanical information, as induced by exercise or any other form of functional activity [94]. The cytoskeleton forms a physically adaptive network to organize cell structure in response to physiologic mechanical signals. Force continuum provided by the tensegrity architecture fills a mechanosensory role by converting mechanical input into a stress throughout the whole cytoskeleton [115]. In this way, cells perceive and adapt to their local physical environment and with RhoA-dependent cytoskeletal restructuring [82]. In MSCs, dynamic mechanical signals including fluid shear [116], substrate strain [86], or low-intensity vibrations [67••] cause rapid activation of FAK, a crucial signaling component of the peripheral focal adhesion, followed by RhoA activation. RhoA-mediated cellular architecture is also responsive to static physical environments: Introducing MSCs to bone-like stiff substrates promotes osteogenic differentiation [53], while softer substrates initiate adipogenic differentiation programs [117, 118].
Mechanical connectivity transmitted from focal adhesions to the cytoskeleton, and then via the LINC complex into the nucleus, is important in regulating the lamin A/C nucleoskeleton, itself implicated in modulating MSC phenotype. MSCs have a more robust lamin A/C network [53] and perhaps increased LINC connectivity compared to pluripotent cells. Lamin A/C increases when MSCs enter the osteogenic lineage [119], a change consistent with the increased cellular stiffness of osteoblasts [120, 121], and lamin A/C overexpression promotes osteogenic differentiation [122]. In contrast, lamin A/C decreases when MSCs undergo adipogenesis [48] and both partial and complete deletion of lamin A/C promotes an adipogenic program in MSCs [123–125]. We recently showed that, similar to lamin A/C dysfunction, disruption of LINC connectivity through knockdown of Sun proteins not only decreases the MSC mechanosensitivity but also accelerates adipogenic commitment [67••].
Wnt/β-catenin signaling is recognized as critical to MCS lineage allocation [126, 127] and skeletal development as mirrored in processes such as fracture healing where Wnt/β-catenin signaling is shown to be strongly present [112]. While it is certain that transmission loss of mechanical cues into the nucleus underwrites such increased adipogenesis, indeed both unloading and microgravity have repeatedly been shown to increase adipogenic phenotype both in vitro and in vivo [128, 113]; LINC function also participates in β-catenin trafficking into the nucleus. Giant nesprins were shown to associate with α-catenin and β-catenin at the nuclear envelope [41], and consistent with increased adipogenesis in LINC-deficient MSCs, deletion of nesprin-1 reduces nuclear β-catenin levels [41]. These findings suggest that perhaps similar to adherent junctions that retain β-catenin at the plasma membrane by forming complexes with α-catenin, the nuclear envelope may serve a similar function to retain β-catenin at—but not inside—the nuclear envelope. Interestingly, Sun1 [129] as well as torsin A in Caenorhabditis elegans [130] have been associated with nucleoporin function that facilitates nuclear β-catenin import. The LINC binding partner emerin also associates with β-catenin but in contrast appears to promote nuclear β-catenin export from the nucleus [40, 43]. These findings suggest that these mechanoresponsive LINC complexes and their binding partners not only participate in β-catenin signaling but that their mechanically adaptive nature may also play a dynamic role in mechanically modulating β-catenin access to nuclear pore complexes to inform MSC phenotype selection.
Indeed, nucleoskeletal structure and its connectivity with cytoskeleton have been shown to be critical in controlling many signaling events important for MSC bone phenotype. Yorkie-homologues Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) alter their nuclear localization [131] in response to mechanical cues to direct MSC fate selection [132]. Loss of YAP/TAZ not only retards osteogenesis and promotes adipogenic phenotype but also results in loss of mechanical control of MSC differentiation [131]. Nuclear translocation of YAP is responsive to dynamic mechanical signals such as substrate strain [133], and as deletion of nesprin-1 inhibits nuclear YAP translocation, this is a LINC-mediated process [134].
Megakaryoblastic leukemia 1 (MKL1) is another transcriptional regulator that alters intracellular localization based on cytoskeletal structure [135]. G-actin binds to MKL1; thus, increased G-actin/F-actin ratio in the cytoplasm has been shown to prevent the MKL1 nuclear entry and de-repress PPARG in the nucleus activity to promote adipogenesis [135]. Interestingly, MKL1 nuclear localization in lamin A/C-deficient Lmna−/− cells display impaired MKL1 translocation into the nucleus [39]. This defect in nuclear MKL1 retention, however, was primarily dependent on emerin’s localization at the nuclear envelope rather than on LINC-mediated nucleo-cytoskeletal connectivity [39]. As such, emerin might affect intranuclear actin dynamics to regulate MKL1 localization rather than controlling MKL1 at a distance in the cytoplasm. Indeed, emerin can cap barbed end of actin filaments and can regulate its polymerization within the nucleus [38].
Although the role of actin within the nucleus is largely uncharted, we recently reported that cytochalasin D-induced actin depolymerization causes rapid influx of G-actin into the nucleus, causing YAP nuclear export, with consequent de-repression of Runx2 activity to accelerate osteogenesis [136•]. While it is not yet clear if actin structure within the nucleus contributes to osteogenic gene expression, we identified filamentous actin inside the nucleus following cytochalasin D treatment [136•]. Polymerized actin within the nucleus has been shown to regulate MKL1/SRF activity [137, 138]. F-actin polymerization in the nucleus due to integrin attachment was shown to be lamin A/C and LINC dependent as depletion of Sun proteins, emerin or lamin A/C repress F-actin formation upon cell spreading [139•]. In this regard, LINC complexes have been implicated as actin nucleation points at the cytoplasm [104]; it is fascinating to consider that LINC complexes, in conjunction with lamin A/C and emerin, might play a role in regulating the nuclear F-actin networks. These findings highlight that actin dynamics have critical roles at either side of the nuclear envelope in regulating MSC lineage allocation and suggest that LINC complexes mechanically and biochemically couple these two compartments to inform MSC fate selection. In this regard, potential mechanical or biomechanical therapies that could reinforce LINC connectivity may present a potentially therapeutic target to combat LINC and nucleoskeleton associated musculoskeletal conditions.
Role of LINC in Aging/Disease
Conditions involving elements of nuclear envelope and nucleoskeleton primarily manifest in mechanosensitive tissues of mesenchymal origin including bone, muscle, and cardiac tissue [140]. Mutations of emerin and lamin are associated with Emery–Dreifuss muscular dystrophy [96] and dilated cardiomyopathy [141]. Importantly, the lamin A/C mutations that cause Hutchinson–Gilford progeria syndrome (HGPS) also show a bone phenotype [142]. The most frequent lamin A/C mutation in HGPS is the nucleotide substitution at position 1824C → T (LmnaΔ50) [143, 144] that results in abnormal farnesylation to generate “progerin” production. Accumulation of progerin at the nuclear rim causes defects in nuclear structure and shape [145]. As opposed to loss of lamin A/C, where the nucleus becomes softer [146], accumulation of progerin on inner nuclear envelope in HGPS patients and to some extent in aged individuals [147, 148] results in a stiffer nucleus [54]. In contrast to the nuclear stiffening caused by mechanical stimuli [44], or osteogenic differentiation [149] in healthy cells, stiffness resulting from progerin accumulation promotes apoptosis and decreases the cell’s ability to sense and respond to environmental mechanical cues [150]. Such aberrant nuclear morphology and stiffness can be rescued by farnesyltransferase inhibitors, which are expected to decrease progerin localization at the inner nuclear envelope [145]. However, improving nuclear stiffness has not been shown to improve cell signaling and apoptosis under mechanical stress [54]. Taken together with evidence that clinical use of the farnesyltransferase inhibitor lonafarnib was associated with positive outcome only in a subset of patients [151], the rescue of nuclear mechanics alone is not likely to reverse global defects seen in HGPS. This underscores that the pathophysiology of HGPS, and similar envelopathies, devolve from a disruption in cell connectivity at the level of the LINC complex.
Even though there has been considerable focus on the isolated defects on the lamin A/C network and its interactions with the transcriptional engine in progeria [55, 141], scientific understanding is shifting to a more integrated paradigm where disconnection of LINC complexes disrupts the flow of physical and molecular information between the two linked cell compartments inside and outside of the nucleus [9, 152]. Highlighting the importance of nuclear actin connections, a wide range of laminopathies that differ in their nuclear mechanics share a common decline in LINC-mediated nuclear cytoplasmic coupling [37]. Importantly, the LmnaL530P/L530P (LmnaΔ9) mutation that closely resembles HGPS [153] and produces a farnesylated form of lamin [154], both Lmn−/− and LmnaΔ9 mouse models revealed a pathological accumulation of Sun-1 in the nuclear envelope and ER membranes; deletion in Sun-1 in these mice partially rescues the bone phenotype [143]. This suggests that loss of LINC function plays an important role in HGPS disease progression.
Critically, healthy aging phenocopies some of the traits of progeria. There is evidence that cells isolated from aged individuals also have low levels of the abnormal progerin and display irregular nuclear shape [148]. In the aging skeleton, adipogenesis is associated with a decline in bone quality [155]. Further, although training can improve skeletal health in the elderly [156], aging bones do not respond adequately to functional loading: Despite daily exercise, bone density is lost and there is increased adiposity within the marrow space. Much like aging phenotype, progeriod mice show bone loss and fat encroachment in the marrow [96]. Critically confirming the close relation between β-catenin mobility and LINC complex [41], LmnaΔ9 mutant progerioid mice (and LmnaΔ50 cells to a lesser extent) show reductions in Wnt activity and cellular β-catenin levels [96]. In this way, lamin A/C haploinsufficient mice with reduced laminA/C levels recapitulate the increased fat, reduced bone, and β-catenin signaling of progeria [157]. Our studies in vitro showed that loss of LINC function alone is able to promote adipogenic phenotype in MSCs [67••]. This suggests that in aging and those diseases that affect the nuclear envelope and nucleoskeleton, the skeletal mechanoresponse becomes less sensitive, and that loss of functional nuclear/cytoskeletal connections may contribute to age related declines in musculoskeletal tissues.
Recent studies also implicate decreased LINC complex components and lamin A/C in carcinogenesis [60•]. Nesprin-2 deletion was associated with reduced fibroblast differentiation along with the dislocation of HP1β (heterochromatin protein 1β) from the nuclear envelope [59]. Importantly, abnormal HP1 isoforms in breast cancer cells have been associated with a poorly differentiated cancer cell phenotype [158] highlighting the importance of cell connectivity in the preservation of health. Interestingly, it has been recently shown that application of low-intensity vibrations can retard tumor progression and increase bone volume in a mouse model of multiple myeloma [159] as well as promoting wound healing response in mice [160]. Although the mechanisms by which low-intensity vibrations improve these outcomes remain undiscovered [161], it is possible that vibration enhanced LINC connectivity or improved nucleoskeletal architecture, as we showed in our in vitro study [67••]. More studies are needed to understand mechanical regulation of nuclear-LINC-cytoskeletal connectivity.
Conclusions
The fact that mechanical signals influence LINC recruitment and its connectivity with the surrounding cytoskeleton suggests that modifications in LINC could make the cell more effective in sensing and responding to mechanical challenges. Such an adaptive response to mechanical challenges would not only affect intracellular mechanical signal transmission but also serve to re-position critical signaling elements such as β-catenin, YAP, and MKL1/SRF to influence cell mechanoresponses. On the other hand, loss of LINC-mediated connectivity plays significant role in how cells perceive their mechanical environment and thus dysfunctional connectivity will contribute to aberrant and subpar cell responses to mechanical signals. Although the mechanisms by which LINC complexes (including the nucleoskeleton) are regulated by mechanical and biochemical factors, or those mechanisms controlling LINC assembly, remain unknown, consideration of mechanical or biochemical strategies that can utilize the adaptive remodeling capabilities of LINC complexes and nucleoskeleton to improve cell connectivity might present novel therapeutic strategies to combat many of the musculoskeletal conditions associated with poor nucleo-cytoskeletal coupling.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Thompson WR, Rubin CT, Rubin J. Mechanical regulation of signaling pathways in bone. Gene. 2012;503(2):179–93.
Wang N, Tytell JD, Ingber DE. Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat Rev Mol Cell Biol. 2009;10(1):75–82.
Styner M et al. Bone marrow fat accumulation accelerated by high fat diet is suppressed by exercise. Bone. 2014;64:39–46.
Case N et al. Mechanical activation of β-catenin regulates phenotype in adult murine marrow-derived mesenchymal stem cells. J Orthop Res. 2010;28(11):1531–8.
Ozcivici E et al. Low-level vibrations retain bone marrow’s osteogenic potential and augment recovery of trabecular bone during reambulation. PLoS One. 2010;5(6), e11178.
Sastry SK, Burridge K. Focal adhesions: a nexus for intracellular signaling and cytoskeletal dynamics. Exp Cell Res. 2000;261(1):25–36.
Fedorchak GR, Kaminski A, Lammerding J. Cellular mechanosensing: getting to the nucleus of it all. Prog Biophys Mol Biol. 2014;115(2–3):76–92.
Crisp M et al. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol. 2006;172(1):41–53.
Luxton GWG, Starr DA. KASHing up with the nucleus: novel functional roles of KASH proteins at the cytoplasmic surface of the nucleus. Curr Opin Cell Biol. 2014;28:69–75.
Martins RP et al. Mechanical regulation of nuclear structure and function. Annu Rev Biomed Eng. 2012;14(1):431–55.
Dauer WT, Worman HJ. The nuclear envelope as a signaling node in development and disease. Dev Cell. 2009;17(5):626–38.
Somech R et al. Nuclear envelopathies: raising the nuclear veil. Pediatr Res. 2005;57(5 Part 2):8R–15R.
Ketema M, Sonnenberg A. Nesprin-3: a versatile connector between the nucleus and the cytoskeleton. Biochem Soc Trans. 2011;39:1719–24.
Padmakumar VC et al. Enaptin, a giant actin-binding protein, is an element of the nuclear membrane and the actin cytoskeleton. Exp Cell Res. 2004;295(2):330–9.
Zhen Y-Y et al. NUANCE, a giant protein connecting the nucleus and actin cytoskeleton. J Cell Sci. 2002;115(15):3207–22.
Wilson MH, Holzbaur EL. Nesprins anchor kinesin-1 motors to the nucleus to drive nuclear distribution in muscle cells. Development. 2015;142(1):218–28.
Roux KJ et al. Nesprin 4 is an outer nuclear membrane protein that can induce kinesin-mediated cell polarization. Proc Natl Acad Sci. 2009;106(7):2194–9.
Simpson JG, Roberts RG. Patterns of evolutionary conservation in the nesprin genes highlight probable functionally important protein domains and isoforms. Biochem Soc Trans. 2008;36:1359–67.
Autore F et al. Large-scale modelling of the divergent spectrin repeats in nesprins: giant modular proteins. PLoS ONE. 2013;8(5), e63633.
Starr DA, Han M. Role of ANC-1 in tethering nuclei to the actin cytoskeleton. Science. 2002;298(5592):406–9.
Lu W et al. Nesprin interchain associations control nuclear size. Cell Mol Life Sci. 2012;69(20):3493–509.
Duong NT et al. Nesprins: tissue-specific expression of epsilon and other short isoforms. PLoS ONE. 2014;9(4), e94380.
Rajgor D et al. Multiple novel nesprin-1 and nesprin-2 variants act as versatile tissue-specific intracellular scaffolds. PLoS ONE. 2012;7(7), e40098.
Dreger M et al. Nuclear envelope proteomics: novel integral membrane proteins of the inner nuclear membrane. Proc Natl Acad Sci. 2001;98(21):11943–8.
Hodzic DM et al. Sun2 is a novel mammalian inner nuclear membrane protein. J Biol Chem. 2004;279(24):25805–12.
Sosa BA, Kutay U, Schwartz TU. Structural insights into LINC complexes. Curr Opin Struct Biol. 2013;23(2):285–91.
Cain NE, Starr DA. SUN proteins and nuclear envelope spacing. Nucleus. 2015;6(1):2–7.
Cain NE et al. The SUN protein UNC-84 is required only in force-bearing cells to maintain nuclear envelope architecture. J Cell Biol. 2014;206(2):163–72.
Sosa BA et al. LINC complexes form by binding of three KASH peptides to the interfaces of trimeric SUN proteins. Cell. 2012;149(5):1035–47.
Rothballer A, Schwartz TU, Kutay U. LINCing complex functions at the nuclear envelope: what the molecular architecture of the LINC complex can reveal about its function. Nucleus. 2013;4(1):29–36.
Haque F et al. Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J Biol Chem. 2010;285(5):3487–98.
Link J et al. Analysis of meiosis in SUN1 deficient mice reveals a distinct role of SUN2 in mammalian meiotic LINC complex formation and function. PLoS Genet. 2014;10(2), e1004099.
Liu Q et al. Functional association of Sun1 with nuclear pore complexes. J Cell Biol. 2007;178(5):785–98.
Manilal S et al. The emery-dreifuss muscular dystrophy protein, emerin, is a nuclear membrane protein. Hum Mol Genet. 1996;5(6):801–8.
Camozzi D et al. Diverse lamin-dependent mechanisms interact to control chromatin dynamics. Nucleus. 2014;5(5):427–40.
Burke B, Stewart CL. Functional architecture of the cell’s nucleus in development, aging, and disease. Curr Top Dev Biol. 2014;109:1–52.
Zwerger M et al. Myopathic lamin mutations impair nuclear stability in cells and tissue and disrupt nucleo-cytoskeletal coupling. Hum Mol Genet. 2013;22(12):2335–49.
Holaska JM, Kowalski AK, Wilson KL. Emerin caps the pointed end of actin filaments: evidence for an actin cortical network at the nuclear inner membrane. PLoS Biol. 2004;2(9), E231.
Ho CY et al. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature. 2013;497(7450):507–11.
Stubenvoll A et al. Attenuation of Wnt/beta-catenin activity reverses enhanced generation of cardiomyocytes and cardiac defects caused by the loss of emerin. Hum Mol Genet. 2015;24(3):802–13.
Neumann S et al. Nesprin-2 interacts with α-catenin and regulates Wnt signaling at the nuclear envelope. J Biol Chem. 2010;285(45):34932–8.
Tilgner K et al. Dynamic complexes of A-type lamins and emerin influence adipogenic capacity of the cell via nucleocytoplasmic distribution of β-catenin. J Cell Sci. 2009;122(3):401–13.
Markiewicz E et al. The inner nuclear membrane protein Emerin regulates [beta]-catenin activity by restricting its accumulation in the nucleus. EMBO J. 2006;25(14):3275–85.
Guilluy C et al. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat Cell Biol. 2014;16(4):376–81.
Ho CY, Lammerding J. Lamins at a glance. J Cell Sci. 2012;125(9):2087–93.
Biamonti G et al. The gene for a novel human lamin maps at a highly transcribed locus of chromosome 19 which replicates at the onset of S-phase. Mol Cell Biol. 1992;12(8):3499–506.
Rober RA, Weber K, Osborn M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development. 1989;105(2):365–78.
Constantinescu D et al. Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem Cells. 2006;24(1):177–85.
Guilak F, Tedrow JR, Burgkart R. Viscoelastic properties of the cell nucleus. Biochem Biophys Res Commun. 2000;269(3):781–6.
Dahl KN et al. The nuclear envelope lamina network has elasticity and a compressibility limit suggestive of a molecular shock absorber. J Cell Sci. 2004;117(20):4779–86.
Lammerding J et al. Lamins A and C but NOT lamin B1 regulate nuclear mechanics. J Biol Chem. 2006;281(35):25768–80.
Pajerowski JD et al. Physical plasticity of the nucleus in stem cell differentiation. Proc Natl Acad Sci U S A. 2007;104(40):15619–24.
Swift J, et al. Nuclear Lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013. 341(6149).
Verstraeten VLRM et al. Increased mechanosensitivity and nuclear stiffness in Hutchinson–Gilford progeria cells: effects of farnesyltransferase inhibitors. Aging Cell. 2008;7(3):383–93.
Gruenbaum Y et al. The nuclear lamina comes of age. Nat Rev Mol Cell Biol. 2005;6(1):21–31.
Lombardi ML et al. The interaction between nesprins and Sun proteins at the nuclear envelope is critical for force transmission between the nucleus and cytoskeleton. J Biol Chem. 2011;286(30):26743–53.
Philip JT, Dahl KN. Nuclear mechanotransduction: response of the lamina to extracellular stress with implications in aging. J Biomech. 2008;41(15):3164–70.
Scaffidi P, Misteli T. Good news in the nuclear envelope: loss of lamin A might be a gain. J Clin Invest. 2006;116(3):632–4.
Rashmi RN et al. The nuclear envelope protein Nesprin-2 has roles in cell proliferation and differentiation during wound healing. Nucleus. 2012;3(2):172–86.
Matsumoto A, et al. Global loss of a nuclear lamina component, lamin A/C, and LINC complex components SUN1, SUN2, and nesprin-2 in breast cancer. Cancer Med. 2015. This work identifies that reduced expression of LINC complex and nuclear lamina components may promote breast cancer progression.
Gaspar-Maia A et al. Open chromatin in pluripotency and reprogramming. Nat Rev Mol Cell Biol. 2011;12(1):36–47.
Eckersley-Maslin MA et al. Lamin A/C is expressed in pluripotent mouse embryonic stem cells. Nucleus. 2013;4(1):53–60.
Chiang FP, Uzer G. Mapping full field deformation of auxetic foams using digital speckle photography. Phys Status Solidi B Basic Solid State Phys. 2008;245(11):2391–4.
Pagliara S et al. Auxetic nuclei in embryonic stem cells exiting pluripotency. Nat Mater. 2014;13(6):638–44.
Ramdas NM, Shivashankar GV. Cytoskeletal control of nuclear morphology and chromatin organization. J Mol Biol. 2015;427(3):695–706.
Ihalainen TO, et al. Differential basal-to-apical accessibility of lamin A/C epitopes in the nuclear lamina regulated by changes in cytoskeletal tension. Nat Mater. 2015. Advance online publication. This study shows that the forces exerted on the nucleus by actin cytoskeleton dictate Lamin A/C basal/apical polarization through LINC complex mediated connectivity.
Uzer G et al. Cell mechanosensitivity to extremely low-magnitude signals is enabled by a LINCed nucleus. Stem Cells. 2015;33(6):2063–76. This work identifies that LINC complexes are essential for sensing low magnitude mechanical signals and that loss of LINC causes accelerated adipogenesis in MSCs. This work further highlights that even though low and high intensity mechanical signals work synergistically to activate FAK/Akt signaling, sites of signal initiation are distinct and result in distinct cytoskeletal adaptations.
Buxboim A et al. Matrix elasticity regulates lamin-A, C phosphorylation and turnover with feedback to actomyosin. Curr Biol. 2014;24(16):1909–17.
Poh Y-C et al. Dynamic force-induced direct dissociation of protein complexes in a nuclear body in living cells. Nat Commun. 2012;3:866.
Chambliss AB, et al. The LINC-anchored actin cap connects the extracellular milieu to the nucleus for ultrafast mechanotransduction. Sci Rep. 2013. 3.
Nagayama K, et al. Estimation of the mechanical connection between apical stress fibers and the nucleus in vascular smooth muscle cells cultured on a substrate. J Biomech. 2014.
Kutscheidt S et al. FHOD1 interaction with nesprin-2G mediates TAN line formation and nuclear movement. Nat Cell Biol. 2014;16(7):708–15.
Versaevel M, et al. Super-resolution microscopy reveals LINC complex recruitment at nuclear indentation sites. Sci Rep. 2014. 4. This work uses high resolution microscopy to elegantly demonstratethat LINC components nesprin and lamin A/C are dynamically recruited to actin stress fibers that produce nuclear indentation.
Padmakumar VC et al. The inner nuclear membrane protein Sun1 mediates the anchorage of Nesprin-2 to the nuclear envelope. J Cell Sci. 2005;118(15):3419–30.
Tanabe LM et al. Primary dystonia: molecules and mechanisms. Nat Rev Neurol. 2009;5(11):598–609.
Jungwirth M et al. The nuclear envelope localization of DYT1 dystonia torsinA-DeltaE requires the SUN1 LINC complex component. BMC Cell Biol. 2011;12(1):24.
Nery FC et al. TorsinA binds the KASH domain of nesprins and participates in linkage between nuclear envelope and cytoskeleton. J Cell Sci. 2008;121(20):3476–86.
Sosa BA et al. How lamina-associated polypeptide 1 (LAP1) activates Torsin. Elife. 2014;3, e03239.
Shin JY et al. Lamina-associated polypeptide-1 interacts with the muscular dystrophy protein emerin and is essential for skeletal muscle maintenance. Dev Cell. 2013;26(6):591–603.
Baker EL, Zaman MH. The biomechanical integrin. J Biomech. 2010;43(1):38–44.
Kanchanawong P et al. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010;468(7323):580–4.
Burridge K, Wittchen ES. The tension mounts: stress fibers as force-generating mechanotransducers. J Cell Biol. 2013;200(1):9–19.
Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Ann Rev Cell Dev Biol. 2005;21(1):247–69.
Riddick N, Ohtani K, Surks HK. Targeting by myosin phosphatase-RhoA interacting protein mediates RhoA/ROCK regulation of myosin phosphatase. J Cell Biochem. 2008;103(4):1158–70.
Sen B et al. mTORC2 regulates mechanically induced cytoskeletal reorganization and lineage selection in marrow-derived mesenchymal stem cells. J Bone Miner Res. 2014;29(1):78–89.
Thompson WR et al. Mechanically activated Fyn utilizes mTORC2 to regulate RhoA and adipogenesis in mesenchymal stem cells. Stem Cells. 2013;31(11):2528–37.
Sen B et al. Mechanically induced focal adhesion assembly amplifies anti-adipogenic pathways in mesenchymal stem cells. Stem Cells. 2011;29(11):1829–36.
Sun Y, Chen CS, Fu J. Forcing stem cells to behave: a biophysical perspective of the cellular microenvironment. Annu Rev Biophys. 2012;41(1):519–42.
Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. 2009;10(1):21–33.
Dalby MJ, Gadegaard N, Oreffo ROC. Harnessing nanotopography and integrin-matrix interactions to influence stem cell fate. Nat Mater. 2014;13(6):558–69.
Rubin J, Rubin C, Jacobs CR. Molecular pathways mediating mechanical signaling in bone. Gene. 2006;367:1–16.
Caille N, Tardy Y, Meister JJ. Assessment of strain field in endothelial cells subjected to uniaxial deformation of their substrate. Ann Biomed Eng. 1998;26(3):409–16.
Chancellor TJ et al. Actomyosin tension exerted on the nucleus through nesprin-1 connections influences endothelial cell adhesion, migration, and cyclic strain-induced reorientation. Biophys J. 2010;99(1):115–23.
Ozcivici E et al. Mechanical signals as anabolic agents in bone. Nat Rev Rheumatol. 2010;6(1):50–9.
Luu YK et al. Mechanical stimulation of mesenchymal stem cell proliferation and differentiation promotes osteogenesis while preventing dietary-induced obesity. J Bone Miner Res. 2009;24(1):50–61.
Hernandez L et al. Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev Cell. 2010;19(3):413–25.
Horn HF et al. The LINC complex is essential for hearing. J Clin Invest. 2013;123(2):740–50.
Rubin C et al. Anabolism. Low mechanical signals strengthen long bones. Nature. 2001;412(6847):603–4.
Gilsanz V et al. Low-level, high-frequency mechanical signals enhance musculoskeletal development of young women with Low BMD. J Bone Miner Res. 2006;21(9):1464–74.
Uzer G et al. Vibration induced osteogenic commitment of mesenchymal stem cells is enhanced by cytoskeletal remodeling but not fluid shear. J Biomech. 2013;46(13):2296–302.
Uzer G et al. Gap junctional communication in osteocytes is amplified by low intensity vibrations in vitro. PLoS ONE. 2014;9:e90840.
Banerjee I et al. Targeted ablation of nesprin 1 and nesprin 2 from murine myocardium results in cardiomyopathy, altered nuclear morphology and inhibition of the biomechanical gene response. PLoS Genet. 2014;10(2), e1004114.
Kim DH, Wirtz D. Cytoskeletal tension induces the polarized architecture of the nucleus. Biomaterials. 2015;48:161–72.
Munter S et al. Actin polymerisation at the cytoplasmic face of eukaryotic nuclei. BMC Cell Biol. 2006;7:23.
Patel MJ et al. Low magnitude and high frequency mechanical loading prevents decreased bone formation responses of 2T3 preosteoblasts. J Cell Biochem. 2009;106(2):306–16.
Sen B et al. Mechanical signal influence on mesenchymal stem cell fate is enhanced by incorporation of refractory periods into the loading regimen. J Biomech. 2011;44(4):593–9.
Francois M, et al. ‘Exercise snacks’ before meals: a novel strategy to improve glycaemic control in individuals with insulin resistance. Diabetologia. 2014: 1–9.
Wolff J. Law of bone remodeling. Berlin: SpringerVerlag; 1986.
Fritton SP, McLeod KJ, Rubin CT. Quantifying the strain history of bone: spatial uniformity and self-similarity of low-magnitude strains. J Biomech. 2000;33(3):317–25.
Goodship AE, Lawes TJ, Rubin CT. Low-magnitude high-frequency mechanical signals accelerate and augment endochondral bone repair: preliminary evidence of efficacy. J Orthop Res. 2009;27(7):922–30.
Morgan EF, De Giacomo A, Gerstenfeld LC. Overview of skeletal repair (fracture healing and its assessment). Methods Mol Biol. 2014;1130:13–31.
Hadjiargyrou M et al. Transcriptional profiling of bone regeneration. Insight into the molecular complexity of wound repair. J Biol Chem. 2002;277(33):30177–82.
Meyers VE et al. RhoA and cytoskeletal disruption mediate reduced osteoblastogenesis and enhanced adipogenesis of human mesenchymal stem cells in modeled microgravity. J Bone Miner Res. 2005;20(10):1858–66.
Moerman EJ et al. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-γ2 transcription factor and TGF-β/BMP signaling pathways. Aging Cell. 2004;3(6):379–89.
Ingber DE, Wang N, Stamenovic D. Tensegrity, cellular biophysics, and the mechanics of living systems. Rep Prog Phys. 2014;77(4):046603.
Riehl BD et al. Fluid-flow-induced mesenchymal stem cell migration: role of focal adhesion kinase and RhoA kinase sensors. J R Soc Interface. 2015;12(104):20141351.
Park JS et al. The effect of matrix stiffness on the differentiation of mesenchymal stem cells in response to TGF-beta. Biomaterials. 2011;32(16):3921–30.
Gobaa S, Hoehnel S, Lutolf MP. Substrate elasticity modulates the responsiveness of mesenchymal stem cells to commitment cues. Integr Biol (Camb). 2015.
Vidal C et al. Role of the nuclear envelope in the pathogenesis of age-related bone loss and osteoporosis. Bonekey Rep. 2012;1:62.
McBeath R et al. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6(4):483–95.
Swift J et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341(6149):1240104.
Bermeo S et al. Lamin A/C acts as an essential factor in mesenchymal stem cell differentiation through the regulation of the dynamics of the Wnt/beta-Catenin pathway. J Cell Biochem. 2015;116(10):2344–53.
Akter R et al. Effect of Lamin A/C knockdown on osteoblast differentiation and function. J Bone Miner Res. 2009;24(2):283–93.
Tong J et al. Lamin A/C deficiency is associated with fat infiltration of muscle and bone. Mech Ageing Dev. 2011;132(11–12):552–9.
Li W et al. Decreased bone formation and osteopenia in lamin a/c-deficient mice. PLoS One. 2011;6(4), e19313.
Sen B et al. Mechanical strain inhibits adipogenesis in mesenchymal stem cells by stimulating a durable beta-catenin signal. Endocrinology. 2008;149(12):6065–75.
Case N et al. β-Catenin levels influence rapid mechanical responses in osteoblasts. J Biol Chem. 2008;283(43):29196–205.
Pan Z et al. Effects of hindlimb unloading on ex vivo growth and osteogenic/adipogenic potentials of bone marrow-derived mesenchymal stem cells in rats. Stem Cells Dev. 2008;17(4):795–804.
Lu W et al. Sun1 forms immobile macromolecular assemblies at the nuclear envelope. Biochim Biophys Acta (BBA) Mol Cell Res. 2008;1783(12):2415–26.
VanGompel MJ et al. A novel function for the Caenorhabditis elegans torsin OOC-5 in nucleoporin localization and nuclear import. Mol Biol Cell. 2015;26(9):1752–63.
Dupont S et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179–83.
Hong J-H et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science. 2005;309(5737):1074–8.
Codelia, Sun G, Irvine KD. Regulation of YAP by mechanical strain through Jnk and Hippo signaling. Curr Biol. 2014;24(17):2012–7.
Driscoll, Tristan P, et al. Cytoskeletal to nuclear strain transfer regulates YAP signaling in mesenchymal stem cells. Biophys J. 108(12): 2783–2793.
Nobusue H, et al. Regulation of MKL1 via actin cytoskeleton dynamics drives adipocyte differentiation. Nat Commun. 2014. 5.
Sen B, et al. Intranuclear actin regulates osteogenesis. stem cells. 2015. This study shows that, as opposed to established dogma in the field that formation of F-actin in cytoplasm drives the MSC osteogenesis, rapid actin depolymerization causes an influx of G-actin into nucleus and activates Runx2 by via YAP nuclear export in a G-actin dependent manner.
Baarlink C, Wang H, Grosse R. Nuclear actin network assembly by formins regulates the SRF coactivator MAL. Science. 2013;340(6134):864–7.
Staus DP et al. Nuclear RhoA signaling regulates MRTF-dependent SMC-specific transcription. Am J Physiol Heart Circ Physiol. 2014;307(3):H379–90.
Plessner M et al. Nuclear F-actin formation and reorganization upon cell spreading. J Biol Chem. 2015;290(18):11209–16. The authors show that actin dynamically polymerizes inside the cell nucleus through an integrin mediated mechanism, and that formation of actin fibers depends on a functional LINC and nuclear envelope structures. Deletion of Sun1/2, emerin or lamin A/C interfere with this process.
Schreiber KH, Kennedy BK, Bad WLG. Nuclear structure and disease. Cell. 2013;152(6):1365–75.
Lammerding J et al. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113(3):370–8.
Lattanzi G. Prelamin A-mediated nuclear envelope dynamics in normal and laminopathic cells. Biochem Soc Trans. 2011;39(6):1698–704.
Chen CY et al. Accumulation of the inner nuclear envelope protein Sun1 is pathogenic in progeric and dystrophic laminopathies. Cell. 2012;149(3):565–77.
Goldman RD et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101(24):8963–8.
Glynn MW, Glover TW. Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum Mol Genet. 2005;14(20):2959–69.
Broers JL et al. Decreased mechanical stiffness in LMNA−/− cells is caused by defective nucleo-cytoskeletal integrity: implications for the development of laminopathies. Hum Mol Genet. 2004;13(21):2567–80.
McClintock D et al. The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS One. 2007;2(12), e1269.
Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312(5776):1059–63.
Darling EM et al. Viscoelastic properties of human mesenchymally-derived stem cells and primary osteoblasts, chondrocytes, and adipocytes. J Biomech. 2008;41(2):454–64.
Tamiello C et al. Soft substrates normalize nuclear morphology and prevent nuclear rupture in fibroblasts from a laminopathy patient with compound heterozygous LMNA mutations. Nucleus. 2013;4(1):61–73.
Gordon LB et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2012;109(41):16666–71.
Fedorchak GR, Kaminski A, Lammerding J. Cellular mechanosensing: getting to the nucleus of it all. Prog Biophys Mol Biol.
Mounkes LC et al. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature. 2003;423(6937):298–301.
Hale CM et al. Dysfunctional connections between the nucleus and the actin and microtubule networks in laminopathic models. Biophys J. 2008;95(11):5462–75.
Mulvihill BM, Prendergast PJ. An algorithm for bone mechanoresponsiveness: implementation to study the effect of patient-specific cell mechanosensitivity on trabecular bone loss. Comput Methods Biomech Biomed Engin. 2008;11(5):443–51.
Bergstrom I et al. Physical training preserves bone mineral density in postmenopausal women with forearm fractures and low bone mineral density. Osteoporos Int. 2008;19(2):177–83.
Duque G, Rivas D. Age-related changes in lamin A/C expression in the osteoarticular system: laminopathies as a potential new aging mechanism. Mech Ageing Dev. 2006;127(4):378–83.
Lee YH et al. HP1beta is a biomarker for breast cancer prognosis and PARP inhibitor therapy. PLoS One. 2015;10(3), e0121207.
Pagnotti GM, et al. Low intensity mechanical signals slow tumor progression and protect bone quantity and quality in a mouse model of multiple myeloma. Bone (New York, N.Y.). 2015. cpa.
Weinheimer-Haus EM et al. Low-intensity vibration improves angiogenesis and wound healing in diabetic mice. PLoS ONE. 2014;9(3), e91355.
Chan ME, Uzer G, Rubin C. The potential benefits and inherent risks of vibration as a non-drug therapy for the prevention and treatment of osteoporosis. Curr Osteoporos Rep. 2013: 1–9.
Acknowledgments
This study was supported by NIH AR056655, AR066616, EB014351, and National Space Biomedical Research Institute PF04304 through NASA NCC 9–58.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Gunes Uzer, Clinton T. Rubin, and Janet Rubin1 declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Molecular Biology of Skeletal Development
Rights and permissions
About this article

Cite this article
Uzer, G., Rubin, C.T. & Rubin, J. Cell Mechanosensitivity Is Enabled by the LINC Nuclear Complex. Curr Mol Bio Rep 2, 36–47 (2016). https://doi.org/10.1007/s40610-016-0032-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40610-016-0032-8