
Abstract
The objective of the study was to develop an amorphous solid dispersion (ASD) for an insoluble compound X by hot melt extrusion (HME) process. The focus was to identify material-sparing approaches to develop bioavailable and stable ASD including scale up of HME process using minimal drug. Mixtures of compound X and polymers with and without surfactants or pH modifiers were evaluated by hot stage microscopy (HSM), polarized light microscopy (PLM), and modulated differential scanning calorimetry (mDSC), which enabled systematic selection of ASD components. Formulation blends of compound X with PVP K12 and PVP VA64 polymers were extruded through a 9-mm twin screw mini-extruder. Physical characterization of extrudates by PLM, XRPD, and mDSC indicated formation of single-phase ASD’s. Accelerated stability testing was performed that allowed rapid selection of stable ASD’s and suitable packaging configurations. Dissolution testing by a discriminating two-step non-sink dissolution method showed 70–80% drug release from prototype ASD’s, which was around twofold higher compared to crystalline tablet formulations. The in vivo pharmacokinetic study in dogs showed that bioavailability from ASD of compound X with PVP VA64 was four times higher compared to crystalline tablet formulations. The HME process was scaled up from lab scale to clinical scale using volumetric scale up approach and scale-independent-specific energy parameter. The present study demonstrated systematic development of ASD dosage form and scale up of HME process to clinical scale using minimal drug (∼500 g), which allowed successful clinical batch manufacture of enabled formulation within 7 months.
Similar content being viewed by others


INTRODUCTION
Hot melt extrusion (HME) is a well-known process in plastic industry to produce products of uniform shape and density and its application dates back to the 1930s (1). HME is recently being used in healthcare industry to manufacture medical devices, amorphous solid dispersion (ASD) to enhance bioavailability of poorly soluble API, targeted release drug delivery systems, implants, bioadhesive films, taste masked products (2–8).
HME has gained increased interest in pharmaceutical industry due to numerous following benefits offered by this technique (6,9):
-
HME is an efficient and solvent-free process hence requires less unit steps compared to other techniques. For example, individual excipients can be directly fed into the extruder barrel wherein efficient mixing of the component occurs by the screw elements; hence, pre-blending of the formulation components is not required. Similarly, to manufacture granules wetting and drying of granules is not required as HME is a solvent-free process.
-
Formulations with high drug loading can be manufactured by HME with desired release profile.
-
Due to intimate mixing of components, good content uniformity can be achieved for very low drug loading.
-
HME allows conversion of API to amorphous form or dispersion of API into very small particles, which can result in enhanced bioavailability, less pill burden and in vivo plasma concentration variability, reduced side effects, and improved patient compliance.
-
HME is a scalable continuous process and product quality can be monitored online.
-
Dosage forms of desired shapes can be easily manufactured by HME process (tablets, granules, pellets, films, implants, etc.)
HME has been successfully applied to enhance solubility of poorly soluble API through formation of solid dispersion of API in a polymeric (or lipid) carrier matrix (3,6–9). Based on the molecular state of the API in the carrier phase, solid dispersions prepared by HME can be categorized as crystalline solid dispersions, amorphous solid dispersions, and solid solutions (10–12). ASD not only offer the inherent free energy benefit but also provide maximum increase in surface area and saturation solubility, which results in higher API solubility and bioavailability. Proper selection of polymeric carrier, thorough evaluation of physicochemical properties, stability, and performance of ASD are key aspects that should be evaluated to ensure successful development of bioavailable dispersion by HME process. Once the formulation components are identified, the effect of HME process conditions on key product attributes should be systematically evaluated to ensure a successful scale up. To develop an ASD dosage form and conduct scale up studies for enablement of phase I clinical batch manufacture, usually more than 2–3 kg API is required if the drug loading is ≥25%. Typically, at the early stage of drug product development, the API synthesis cost is high and very limited API is available to develop clinical formulation. Hence, there is a need to identify and implement material-sparing methodologies to enable development of bioavailable ASD and successful scale up of process for manufacture of ASD using minimal API. Currently, only few studies in literature have studied and discussed material-sparing methodologies to develop ASD (13–17). These literature reports are very informative but describe only some of the aspects of ASD development. Reports on full systematic development of ASD from mini-scale to clinical scale using material-sparing approaches are not widely published. The aim of the current study was to identify and implement material-sparing approaches to systematically develop bioavailable as well as stable ASD for an insoluble compound X and conduct scale up of HME process from mini-scale to clinical scale using minimal API, resources, and time.
In this investigative study, a weakly basic drug (referred in this article as compound X), which belongs to BCS class II category was used. The compound X has poor aqueous solubility (intrinsic solubility of 18 μg/ml), moderate hydrophobicity (log P of 2.6) and pK a of 2.5 (shows pH-dependent solubility). The compound X has melting point of ∼209°C, glass transition temperature (T g) of 77°C and decomposes above 275°C. During pre-formulation studies, it was observed that compound X is difficult to convert to amorphous form, which may be due to high crystal lattice energy. It was also observed that if amorphous form of compound X was formed, it readily converted back to crystalline form due to poor physical stability. At the required clinical dose strengths of 100–200 mg, the dose numbers were above 20; hence, there was a need to develop an enabled formulation such as ASD by a suitable process to achieve good exposure in clinical trials.
MATERIALS AND METHODS
Materials
Compound X was supplied by the Chemical Development Department of Boehringer Ingelheim Pharmaceuticals, Inc. (Ridgefield, CT, USA). PVP VA64 (Kollidon® VA64), PVP K12 (Kollidon® K12), and Crospovidone (Kollidon® CL) were obtained from BASF (Ludwigshafen, Germany). HPMC E5 was obtained from the Dow Chemicals (Redmond, WA) and HPMCAS LF was obtained from Shin-Etsu (Totowa, NJ). Sodium dodecyl sulfate (SDS) was purchased from Spectrum Chemicals (New Brunswick, NJ). Tartaric acid was purchased from Mutchler Inc. (Harrington Park, NJ). Microcrystalline cellulose (Avicel® PH 102) was obtained from FMC BioPolymer (Philadelphia, PA). Colloidal silicon dioxide (Aerosil® 200 P) was purchased from Evonik Degussa Corporation (Parsippany, NJ). Magnesium stearate was purchased from Mallinckrodt (Phillipsburg, NJ). Other pharmaceutical excipients of compendial grade were used as received.
Methods
Calculation of Solubility Parameters
The solubility parameter (δ) of compound X and commonly used HME polymers was determined by HSPiP software version 4.1 (Hansen-Solubility, UK), which is based on Hansen solubility parameter estimation approach (18). The chemical structure of the compound X and polymers were drawn and solubility parameters were determined by Van Krevelen and Hoftyzer’s group contribution method using the following equation (19):
Where, δ is the total solubility parameter, δd is contribution from dispersion forces, δp is contribution from polar forces, and δh is contribution from hydrogen bonding. For polymers, the solubility parameters were determined based on the single repeating monomer unit and the average molecular weight. The differences (∆δ) between the solubility parameter values of the compound X and polymers were calculated.
Hot Stage Microscopy (HSM)
An Olympus BX51 optical microscope (Olympus Optical Company, Westmont, IL) equipped with polarizing attachment (model: BXP) and controlled by Mettler Toledo FP82HT hot stage and FP90 Central Processor was used to conduct hot stage evaluation of blends. Spot™ Flex camera system (Diagnostic Instruments Inc., USA) was used to take polarized light microscopy pictures throughout the entire hot stage evaluation. For the hot stage evaluation, ∼15 mg of intimately mixed blend was placed onto a glass slide and heated at 5°C/min. HSM was used to observe the crystal form change or the crystal dissolution processes under controlled temperature by polarized light microscopy. To assess, whether the components formed a single or multi-phase system upon heating, all the samples were heated above the melting point of the compound X. Compound X tends to dissolve into the molten mass of the miscible polymer prior to the melting point. This molten mass was subsequently cooled back to room temperature to evaluate if the dissolved compound X crystallizes out upon cooling to ambient conditions.
Hot Melt Extrusion at Lab Scale (9 mm)
A physical mixture of compound X with polymer and additional components such as acidifier or surfactant was prepared by blending in a Turbula mixer for 5 min. Early-stage hot melt extrusion formulations were prepared by extruding the physical mixture through a 9-mm mini-extruder, which was fabricated at Boehringer Ingelheim in cooperation with ThreeTec (ThreeTec, Seon, Switzerland). The 9-mm extruder was equipped with co-rotating twin screws and heated barrel with two zones. The extruder was heated to desired temperature with thermostatic control at the front and rear end of barrel to maintain a desired barrel temperature. The system was allowed to heat soak for ∼15 min. The twin screws were rotated to a desired speed and the powder blend was added in small amounts. The screw profile consisted of only conveying screw elements. All the glassy material exiting the extruder through a 1-mm round die opening were collected onto an aluminum foil and allowed to cool to room temperature and then stored in a sealed aluminum pouch to keep them moisture free.
Hot Melt Extrusion at Large Scale (16 and 18 mm)
The scale up of HME process was conducted using 16- and 18-mm extruders (Leistritz, Somerville, NJ), equipped with intermeshing co-rotating twin screws. Leistritz’s 16-mm nano-extruder consisted of four heating zones and a volumetric dosing feeder, which was used to feed the physical blend from the bottom into the extruder. During extrusion, zones 1–3 were set at 180°C and zone 4 (near die region) was set at 165°C. The screw profile consisted of various types of conveying elements and a set of kneading elements of 30° and 60° angle to facilitate dissolution of compound X in molten polymer (Fig. 1b). The molten dispersion was passed through a 2-mm round die opening to obtain round extrudates. The extrusion process was monitored by recording melt temperature, melt pressure, and torque values throughout the extrusion run (Table I).

Screw profile of Leistritz’s a micro 18-mm and b nano 16-mm extruder
The 18-mm extruder consisted of seven heating zones, co-rotating twin screws, and a die plate with 3-mm round die opening. All heating zones were maintained at 180°C during the extrusion run. The screw profile consisted of various types of conveying elements and a set of kneading elements of 30°, 60°, and 90° angle to provide adequate mechanical shear energy for formation of ASD (Fig. 1a). The 18-mm extruder was equipped with a twin-screw gravimetric dosing feeder (K-Tron, Pitman, NJ). The extrusion process was monitored by recording melt temperature, melt pressure, and torque values throughout the extrusion run. The extrusion process conditions and additional system details of each scale extruder are shown in Table I.
The cooled extrudates were milled by passing through 30 mesh screen in a quadro co-mill (Waterloo, Canada) at 500 rpm. Milled extrudates were stored in a sealed aluminum pouch to keep them moisture free.
The residence time was determined while conducting extrusion in 16- and 18-mm extruder using red iron oxide as tracer. Around 5 mg of red iron oxide was mixed with 5 mg of blend and added to the extruder after the extrusion process reached equilibrium. The onset as well as offset time for tracer appearance in extrudate was determined visually while conducting extrusion at various process conditions in 16- and 18-mm extruder.
Characterization of Solid Dispersions
Assay and Purity Testing
Quantitative assay and purity analysis of samples was done using an gradient HPLC method where the eluent (A) comprised of 45 mM ammonium hexafluorophosphate in water/methanol 95/5 (v/v) and eluent (B) comprised of methanol/water 95/5 (v/v). The analytical column Atlantis T3 C18, 3 μm, 150 × 4.6 mm was operated at 40°C with a flow rate of 1.0 ml/min and UV detection at 250 nm. The injection volume was 12 μl and the data acquisition time was 57 min.
X-Ray Powder Diffraction (XRPD)
XRPD analysis was performed at ambient temperature using a Bruker AXS X-Ray Powder Diffractometer Model D8 Advance (Karlsruhe, Germany), at 40 mA and 40 KV with CuKα radiation (1.54 Å) in parallel beam mode utilizing a scintillation detector. Samples were scanned over a range of 2θ values from 3° to 35° with a step size of 0.05° (2θ) and a counting time of 4 or 0.6 s. A 1-mm divergence slit was used with the incident beam along with 0.12-mm soller slits in the diffracted beam path. A sodium iodide scintillation detector was used.
Polarized Light Microscopy (PLM)
Polarized light microscopy was performed using an Olympus BX51 Polarizing Microscope (Olympus Optical Company, Westmont, IL) with objective of 20× and ocular magnification of 10×. A small amount of sample was placed on the glass slide followed by addition of a drop of oil, which was covered with a cover slip and then examined for birefringence. Sample analysis was done by using SPOT advanced software.
Thermal Analysis (mDSC)
Modulated differential scanning calorimetry (mDSC) was performed on a DSC Q1000 (TA instruments, New Castle, DE). Data analysis was done using Universal Analysis 2000 thermal analysis software (TA instruments, New Castle, DE). For mDSC, samples of 2–4 mg were weighed and placed in aluminum crimped pans. The samples were equilibrated at 20°C and then heating-cooling-heating cycle was performed. During the heating cycle, the mDSC parameters were modulated at 0.636°C every 60 s with heating rates of 2°C/min from 20 to 230°C followed by holding the sample isothermally for 1 min. During the cooling cycle, the samples were cooled at 2°C/min to −10°C followed by holding the sample isothermally for 1 min and then the samples were subjected to heating cycle. All measurements were performed under nitrogen purge.
Stability Study
The physical stability of solid dispersions was assessed by placing the samples under stress stability condition at 40°C/75% RH in open vials for 7 days to allow for rapid selection of stable amorphous solid dispersion. The accelerated stability on selected amorphous dispersion and capsule formulation was conducted in polypropylene bottle and aluminum pouch at 40°C/75% RH to select appropriate packaging system for long-term storage. The long-term ICH stability study was conducted in selected packaging system to identify adequate shelf life of the amorphous dosage form. At each time point, PLM analysis was conducted to monitor for potential crystallization of the solid dispersions. XRD, mDSC, and dissolution testing were conducted on selected samples.
Dissolution Testing
Dissolution of solid dispersions was performed using Leap OD Lite UV Fiber optic system (North Brunswick, NJ). A two-step non-sink dissolution method was developed to assess the performance of amorphous solid dispersions and crystalline dosage forms. The samples were suspended in 40-mesh basket. The rotation speed was 100 rpm. In the first step, dissolution testing was conducted in 300 ml of simulated gastric fluid at pH = 2 for 30 min, and in the second step, 600 ml phosphate buffer containing 0.15% SDS was added to make a final 900 ml volume of pH = 6.5 media containing 0.1% SDS. The amount of drug released was monitored in the combined media at 330 nm.
In Vivo Testing
The relative bioavailability of four oral solid formulations compared to an oral solution of compound X was studied in six male beagle dogs (protocol # 08-922-E). Animals were fasted overnight before dosing and fed 4 h after dosing. One hour prior to dosing, each dog was pretreated with pentagastrin at 6 μg/kg IM in order to decrease gastric pH to levels similar to humans. In addition, each dog was pretreated with acepromazine maleate at 0.20 mg/kg IM 30 min prior to dosing in order to decrease nausea/vomiting. Animals were administered 100 mg of compound X as an oral solution (10 mg/ml) or a solid dosage formulation followed by 50 ml of water via gavage. Blood samples were obtained pre-dose and at 0.33, 0.67, 1, 1.5, 2, 3, 4, 6, 8, 24, 30, 48, 54, and 72 h post-dose. Each treatment was separated by a 2-week washout period due to the long half-life of compound X in dogs. Plasma concentrations of compound X were analyzed using a validated liquid chromatography-tandem mass spectrometry. Non-compartmental pharmacokinetic parameters based on individual plasma concentration-time profiles were calculated using Kinetica (version 4.4.1, Thermo Scientific, Philadelphia, PA). The linear trapezoidal method was used for AUC0–72 calculations.
RESULTS AND DISCUSSION
Systematic Identification of Solid Dispersion Components
The ideal type of ASD for increasing dissolution is a glass solution, in which the amorphous API has a lower thermodynamic barrier to dissolution together with a maximally reduced particle size, which can be developed by HME process through a careful selection of polymeric carrier. The selection of an appropriate hydrophilic polymer/carrier is crucial to enable development of a bioavailable and stable ASD as well as to successfully conduct the extrusion process. The ideal polymer should readily dissolve the API in its matrix to form amorphous dispersion without causing degradation of API and be processable at the applied processing conditions. In addition, the selected polymer needs to minimize molecular mobility of the API within the matrix through various types of interaction with API molecules (i.e., hydrogen bonds or electrostatic) and/or creating tortuous matrix to achieve long-term storage physical stability of ASD. Use of systematic approaches to develop solid dispersions can potentially reduce the number of experiments and API usage at early stage of development. Instead of solely relying on chemometric tools or mixing API and polymers randomly in more of a “screening mode” followed by extrusion, polymers for evaluation should be selected based upon API-polymer physicochemical properties such as solubility parameters, hydrogen bonding, and thermal properties (see flowchart in Fig. 2). In the present study, a combination of chemometric tool such as solubility parameter, hydrogen bonding potential of various polymers with compound X, and material-sparing experimental techniques such as HSM coupled with PLM and mDSC were used to identify suitable polymers to develop ASD of compound X using minimal API and resources.

Flowchart to develop amorphous dispersions for compound X by HME process using material-sparing approaches
Solubility parameters (δ) are widely used to identify miscible solvent-solute combinations in chemical and polymer applications. The rule of “like dissolves like” applies, i.e., two materials with similar solubility parameters are expected to be miscible. In general, a combination of API and polymer with a difference in solubility parameter (∆δ) of 7.0 MPa1/2 or less is considered miscible (20,21). The solubility parameters (δ) of compound X and commonly used HME polymers as well as the difference between the solubility parameter of compound X and HME polymer (∆δ) are shown in Table II. As shown in Table II, a difference in solubility parameter of (∆δ) ≤7.0 MPa1/2 was observed between compound X and PVP, PVP VA64, HPMC, and HPMCAS polymers; hence, based on solubility parameter theory, these polymers were selected for further evaluation (20,21).
The compound X has hydrogen bond donor group as shown in Fig. 3. Hence, compound X was estimated to form hydrogen bonding with polymers containing hydrogen bond acceptor groups such as PVP, PVP VA64, and HPMCAS, which could result in formation of a stable amorphous dispersion.

Generic structure of compound X
The miscibility of compound X with selected polymers was determined by HSM at varying ratios of 1:1, 1:2, and 1:3. The HSM analysis showed that compound X is completely miscible in PVP VA64 polymer at 1:2 or 1:3 ratio around 190–200°C and in PVP polymer only at 1:3 ratio around 195–205°C. Moreover, no recrystallization of dissolved compound X was observed upon cooling back to room temperature. The compound X was only partially miscible in HPMC and HPMCAS polymer even at higher ratios of 1:2 or 1:3 as some non-strain related birefringence was observed at temperatures >200°C during HSM evaluation, which could be due to presence of small amount of crystalline compound X (Fig. 4). Figure 4 shows the HSM analysis images of compound X with PVP, PVP VA64, HPMCAS, and HPMC polymers of selected blends that were further analyzed by mDSC. The HSM observations were confirmed by mDSC analysis of selected blends. As noted by Zecevic and Wagner (17), the use of HSM method allowed detection of mono- or bi-phasic systems including amorphous or crystalline state of each dispersion component. Moreover, use of slow-heating ramp during HSM evaluation allowed close monitoring of rate and extent of dissolution of compound X in polymer matrices to enable selection of appropriate polymers and estimation of processing temperature of melt in the extruder (17).

Polarized light microscopy images during hot stage microscopy evaluation of a compound X:PVP VA64 (1:2), b compound X:PVP K12 (1:3), c compound X:HPMC (1:2), and d compound X:HPMCAS LF (1:2) blends
The use of mDSC methodology allows estimation of T g, detection of single and multiphase system (based on presence of single or multiple T g), and determination of changes to melting point of the API, which is indicative of potential interaction between API and polymer; hence, it has been used by several researchers successfully to facilitate polymer selection (14,16,17,21). The mDSC analysis results of selected compound X and polymer blends are shown in Fig. 5. The mDSC analysis showed presence of a single T g and no melting peak for compound X:PVP VA64 (1:2) and compound X:PVP (1:3) blends, which indicated that compound X is miscible in these polymer system and forms a single-phase system. The compound X was only partially miscible in HPMC and HPMCAS polymer at 1:2 ratio as small melting peak was observed by mDSC analysis (Fig. 5).

DSC thermograms of compound X and blends of compound X and HME polymers
The HSM evaluation indicated that miscibility of compound X in HPMC and HPMCAS is limited and >200°C extrusion temperature would be required to manufacture ASD using these polymers. Conducting extrusion above 200°C is generally not preferred as it could result in degradation of these polymers. Based on HSM and mDSC analysis the compound X:PVP VA64 (1:2) and compound X:PVP (1:3) formulations were selected for further evaluation due to formation of a single-phase ASD at an acceptable extrusion temperature range (180–195°C). Overall, using material-sparing methodologies such as solubility parameter, HSM, PLM, and mDSC, suitable polymers to prepare ASD of compound X by HME process could be identified in systematic way using only few milligrams of API as outlined in Fig. 2.
Development of HME Prototype Amorphous Solid Dispersions
Based on systematic evaluation (see “Systematic Identification of Solid Dispersion Components” section) the compound X:PVP VA64 (1:2) and compound X:PVP (1:3) formulation blends were selected to develop prototype ASD by HME process. To develop bioavailable, stable, and robust prototype ASD’s of compound X factors such as ease of manufacturability, dissolution enhancement, solid-state characteristics, chemical stability, and physical stability were considered. The development of prototype ASD of compound X with each selected polymer is described in detail in the following sections.
PVP VA64 Containing Amorphous Solid Dispersions
The extrusion of compound X:PVP VA64 blend (formulation 3a) was conducted on lab scale 9-mm mini-extruder at a batch size of 12 g. The extrusion temperature was around 190°C, which was similar to the extrusion temperature identified by HSM evaluation of this blend. The processability of the batch through the extruder was acceptable and no significant cleaning efforts were required after the extrusion run. The extrudates were transparent light yellow, brittle, and easy to mill. The formation of ASD was confirmed by PLM, XRD, and mDSC analysis (Table III). A single T g of ∼94°C was observed by mDSC, no biregringence was observed by PLM, and no crystalline peaks were observed in X-ray diffractogram. The dissolution testing of milled extrudates of formulation 3a was conducted by a two-step non-sink dissolution method and the dissolution profile is shown in Fig. 6a. Dissolution analysis indicated that the total amount of drug released from physical mixture at the end of dissolution testing was ∼35% (shown as solid line for representation in Fig. 6a, b). The amount of drug released from PVP VA64 containing ASD (formulation 3a) at the end of acid stage of dissolution testing was 70–75%, but precipitation of compound X was observed in the buffer stage at later time points of dissolution testing (Fig. 6a), which was due to low solubility of compound X in buffer medium (representing intestinal pH of 6.5) compared to acidic medium (representing gastric pH of 2). The dissolution results indicated that including only PVP VA64 in ASD of compound X may not be sufficient to inhibit the precipitation of compound X when the dispersion will transit from gastric environment to intestinal environment in vivo. To minimize precipitation of compound X, the impact of SDS as solubilizer was evaluated by adding intra-granularly during extrusion (formulation 3e—compound X:PVP VA64:SDS in 1:2:0.5 ratio) and extra-granularly in gelatine capsule along with milled extrudates (formulation 3f). It was observed that addition of SDS extra-granularly resulted in maintenance of supersaturation of compound X during buffer stage of dissolution testing (see dissolution curve for formulation 3f in Fig. 6a). Also, extra-granular addition of SDS to prepare formulation 3f did not negatively impact the physical and chemical stability of the amorphous dispersion (see ICH stability study discussion section for capsule formulation 3f). When SDS was added during extrusion process (formulation 3e), it resulted in a lowering of T g of ASD compared to binary ASD containing compound X and only PVP VA64 (formulation 3a), suggesting that the surfactant is plasticizing the polymer matrix, which resulted in a lower extrusion temperature of 165–170°C and better processability (Table III). Use of surfactants as plasticizers in preparing solid dispersions of poorly soluble drug has also been reported by Ghebremeskel et al. (22). The formation of ASD for formulation 3e was confirmed by PLM, XRPD, and mDSC analysis (Table III). The dissolution analysis indicated much higher drug release (80–90%) from the ternary ASD of formulation 3e and maintenance of supersaturation compared to binary ASD of formulation 3a (Fig. 6a). However, much higher level of impurities (∼9%) and lower physically stability at accelerated stability conditions was observed for SDS containing ternary ASD (formulation 3e) compared to the binary ASD of compound X containing only PVP VA64 (formulation 3a) as shown in Table III. Hence, inclusion of SDS during extrusion was not considered a suitable option for further evaluation.

Dissolution of compound X:PVP VA64 amorphous solid dispersion a with and without SDS and b with and without tartaric acid
In an effort to improve dissolution performance and maintain supersaturation for PVP VA64 containing ASD of compound X, inclusion of various types of acidifiers (fumaric acid, citric acid, tartaric acid, and sodium acid sulfate) during extrusion was evaluated by HSM at various ratios (data not shown). The HSM evaluation indicated that the miscibility of compound X in PVP VA64 polymer was enhanced in the presence of each studied acidifier and occurred at lower temperature of 150–175°C compared to binary mixture of compound X with PVP VA64. Since tartaric acid has lower melting temperature and is less hygroscopic compared to other studied acidifiers, it was selected for further evaluation to prepare ternary ASD by HME. When tartaric acid was added during extrusion process (compound X:PVP VA64:tartaric acid—1:1.5:2) it resulted in lower T g value of 81°C, lower extrusion temperature of 165–170°C, and formation of an ASD, which was confirmed by PLM, XRD, and mDSC analysis (Table III). The dissolution analysis showed much higher drug release (90–95%) from tartaric acid containing ternary ASD (formulation 5b) and maintenance of supersaturation compared to binary ASD of compound X without tartaric acid (formulation 3a) as shown in Fig. 6b. Enhancement in percent drug release could be due to better maintenance of micro-environmental pH, which results in higher solubility by inclusion of tartaric acid or formation of an in situ compound X-tartaric acid co-crystal solid dispersion. Similar results of dissolution enhancement due to formation of in situ co-crystals during hot melt extrusion of mixture of either API and coformer or API, coformer, and polymer carrier have been reported by other researchers (23,24). In the present study, however, much higher levels of impurities (∼10%) were observed for tartaric acid containing ternary ASD (formulation 5b), which could be related to degradation of tartaric acid as the tartaric acid degradation temperature of 175°C was close to the extrusion temperature of 165–170°C. Also, physical stability was compromised by inclusion of tartaric acid compared to the binary ASD containing only PVP VA64 (Table III). Hence, inclusion of tartaric acid during extrusion was not considered a viable option to further develop a prototype ASD.
PVP Containing Amorphous Solid Dispersions
The PVP of K12 grade was selected to prepare ASD of compound X by HME process as it can be extruded at lower extrusion temperature due to lower T g value compared to PVP K30 or K90 grade and has good thermal stability as well as lower viscosity (25). The HSM analysis indicated that compound X is completely miscible in PVP K12 polymer at 1:3 ratio around 195–205°C. The extrusion of compound X:PVP (1:3) formulation blend (formulation 7a) was successfully conducted using a 9-mm extruder at 195°C, which resulted in formation of transparent yellow extrudate. The processability of the batch through the extruder was acceptable and no significant cleaning efforts were required after the extrusion run. The extrudates were brittle and easy to mill. The milled extrudates were amorphous by PLM, mDSC, and XRD analysis (Table III). A single T g of ∼75°C was observed by mDSC. The T g value of PVP K12 containing ASD (formulation 7a) was lower compared to T g value OF ∼94°C for PVP VA64 containing ASD (formulation 3a) as shown in Table III. The PVP K12 ASD was more hygroscopic compared to PVP VA64 ASD (obtained around 5% moisture content in PVP K12 ASD and 2.5% moisture content in PVP VA64 ASD by thermo gravimetric analysis). The presence of extra water in the PVP K12 ASD might have resulted in much lower T g value compared to PVP VA64 ASD. When the PVP K12 ASD was dried to low moisture content and DSC analysis was reconducted, the T g value for PVP K12 ASD increased to 93°C, confirming that absorbed water can lower the T g of an amorphous dispersion due to plasticizing effect similar to the observations made by other researchers (26,27). The dissolution analysis of PVP K12 ASD indicated an increase in compound X release to 70% in simulated intestinal buffer compared to crystalline tablet formulation (Fig. 7a). The drug release increased gradually from acid phase to intestinal buffer phase and no precipitation was observed until the end of dissolution testing. The PVP K12 ASD disintegrated slowly during dissolution testing and also some gelling of the dispersion was observed compared to PVP VA64 ASD, which might have resulted in slow drug release from PVP K12 ASD and also contributed towards preventing precipitation during dissolution testing. The impurity analysis indicated very low level of total impurities (0.2%) (Table III). Impact of addition of SDS and acidifiers during extrusion to the PVP K12 containing blend of compound X was evaluated but either the solid dispersion was not completely amorphous or dissolution enhancement was not observed. In addition, an increase in level of impurities was observed; hence, ternary prototypes for PVP containing formulations of compound X were not pursued further.

Amorphous and crystalline formulations of compound X tested in dogs at 100 mg dose. a Dissolution profiles. b Mean plasma profiles
Overall, ASD’s of compound X could be successfully developed by HME process. Based on criteria such as ease of manufacturability, enhancement in dissolution, physical stability, and low level of impurities during processing, ASD formulation containing PVP VA64 with 10% SDS added extra-granularly (formulation 3f) and ASD formulation containing PVP K12 (formulation 7a) were selected for dog PK study.
In Vivo Assessment of Amorphous Solid Dispersions
The milled extrudates of selected ASD formulations were filled in gelatin capsule along with other extra-granular ingredients to develop prototype ASD capsule dosage form of compound X for in vivo assessment in dogs. In addition, two conventional tablet formulations of crystalline compound X with and without SDS as solubilizer were also developed for in vivo assessment in dogs. The composition of formulations tested in dogs is shown in Table IV. The in vitro performance of the ASD capsules and tablet formulations selected for dog pharmacokinetic study was evaluated using a two-stage non-sink dissolution method. The dissolution profiles in Fig. 7a show that PVP VA64 containing ASD (formulation 3f) achieved higher dissolution at early time points and its supersaturation was maintained in the second step of dissolution. The PVP K12 containing ASD (formulation 7a) showed slower release in steps 1 and 2 of dissolution medium, but at the end of 90 min, the percent drug release was the same as that of PVP VA64 containing ASD of compound X. The overall percent drug release at 90 min from the ASD prototypes was higher compared to crystalline prototype tablets with and without SDS as solubilizer (Fig. 7a). These prototype solid dosage forms were tested in dog PK study in a randomized crossover design using a solution formulation as the reference at 100 mg dose. The plasma profiles of each formulation of compound X tested in dogs are shown in Fig. 7b and the pharmacokinetics parameters are shown in Table VThe rank order of the bioavailability relative to the solution formulation was as follows: PVP VA64 containing ASD (80.6%) > PVP containing ASD (57.6%) > crystalline tablet with SDS (44.3%) > crystalline tablet without SDS (22.7%) (Fig. 7b and Table V). Based on the in vivo performance of compound X formulations, the dissolution profiles obtained by a two-stage dissolution method were carefully reviewed to understand key dissolution characteristics that might have affected the bioavailability from these formulations (Fig. 7a). The lower in vivo performance of PVP K12 containing ASD (formulation 7a) compared to PVP VA64 containing ASD of compound X suggests that fast and higher release of drug in stomach environment may be required in addition to higher release in intestinal environment at early time points (see the first 30–40 min profile in Fig. 7a) to achieve better bioavailability from an ASD formulation of compound X. Also, higher bioavailability from crystalline tablet with SDS compared to crystalline tablet without SDS is suggesting that higher drug release in stomach environment may be required (see the first 30 min profile in Fig. 7a) to achieve better bioavailability. The in vivo results suggest that the developed two-step non-sink dissolution method has formulation discriminating power and helps to identify the formulation and release characteristics needed to achieve high bioavailability of compound X.
Overall, the in vivo assessment of prototype ASD and crystalline tablet formulations of compound X showed a four-times higher exposure for PVP VA64 containing ASD compared to crystalline tablet formulation.
Chemical and Physical Stability Assessment of Amorphous Solid Dispersions
Although, conversion of API to the high-energy amorphous state can be effective in bioavailability enhancement, it is often associated with physical stability risks. The high-energy amorphous form tends to transform into thermodynamically more stable crystalline form overtime ( 28–30). One common approach to kinetically stabilize the high-energy amorphous form is to form an ASD using a suitable polymeric carrier, which can interact with API and decrease the mobility of the amorphous API molecules. In the case of PVP VA64 containing dispersion of compound X, the hydrogen bonding between the carbonyl group of the polymer and NH group of the API was observed by FT-IR and Raman analysis as reported in our earlier published article (31), suggesting that the selected ASD might be physically stable. Other factors such as T g, viscosity, plasticization, storage temperature, and hygroscopicity of the amorphous system also play an important role in determining kinetic stability and development of high-energy ASD’s.
The T g of a dispersion system generally provides a first handle when assessing stability of homogeneously mixed ASD. As an approximation, a difference of 50°C between the storage temperature and T g of an ASD provides initial support to advance amorphous formulation approach to further in development (32). The T g of PVP VA64 containing ASD was around 94°C (Table III), which is >50°C above the storage temperature; hence, good physical stability of the PVP VA64 containing ASD of compound X was expected. Whereas the T g of PVP K12 containing ASD was around 75°C due to higher hygroscopicity of PVP K12 containing ASD, thereby suggesting lower physical stability compared to PVP VA64 containing ASD of compound X (Table III). The stress stability testing of milled extrudates was conducted in open dish at 40°C/75% RH condition for 7 days for both PVP VA64 and PVP K12 containing ASD formulation and stability samples were tested by PLM and XRD. No recrystallization of compound X was observed for PVP VA64 containing ASD whereas some recrystallization of compound X was observed by PLM for PVP K12 containing ASD at 7 days (Fig. 8). The PVP K12 containing ASD was found to be more hygroscopic compared to PVP VA64 containing ASD. A bsorption of excess moisture on stability can negatively impact the physical stability of ASD as the absorbed water can plasticize the system and further reduce the T g of an ASD (26,27). Thus, due to lower bioavailability and physical stability, the PVP K12 containing ASD of compound X was not further studied.

PLM images of stability samples of HME amorphous solid dispersions of compound X
Further stability assessment of PVP VA64 containing ASD of compound X was conducted in following two stages:
-
Packaging screening study—the accelerated stability study of milled extrudates and capsule formulation at 40°C/75% RH condition in various packaging configuration (polypropylene bottle without desiccant and aluminum pouch) was evaluated. The accelerated stability study results showed some recrystallization of compound X when stored in a polypropylene bottle at 40°C/75% RH condition in 4 weeks by PLM whereas no recrystallization was observed in aluminum pouch at the same time point (data not shown). Based on the findings from preliminary packaging screening study, protective closed packaged system such as aluminum blister card was selected to store PVP VA64 containing ASD in hard gelatin capsule for long-term stability.
-
Long-term ICH stability study of milled extrudates and capsule formulation 3f in aluminum blister—the long term ICH stability study was conducted at 25°C/60% RH and 40°C/75% RH condition. On long-term ICH stability, no change in physical stability, chemical stability, and dissolution performance was observed for PVP VA64 containing ASD (formulation 3f) packaged in aluminum blister. There was no change in impurity levels of ASD at ICH stability conditions (data not shown). There was no appearance of crystalline peak of compound X by XRD until 6 months at 40°C/75% RH condition in aluminum blister (Fig. 9a). The dissolution performance of initial and stability samples was similar by a two-stage non-sink dissolution method (Fig. 9b). A slightly higher drug release during acid phase was observed for 3 and 6 months 40°C/75% RH samples, which was attributed to be within dissolution variability as high variability (∼8–12%) was observed at early time points during gelatin capsule dissolution of formulation 3f.
Fig. 9 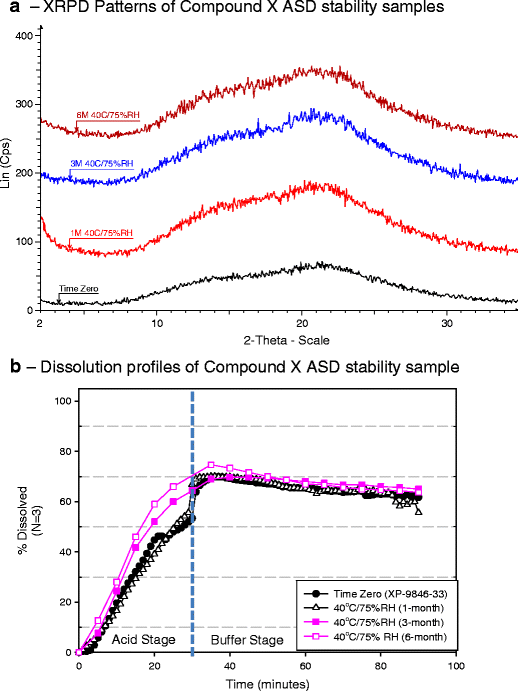
Compound X:PVP VA64 (1:2) ASD samples packaged in aluminum blister—evaluation on long-term ICH stability study by a XRPD and b dissolution profiles

Scale Up of HME Process to Clinical Scale
In the production of pharmaceutical formulations, which require homogeneous mixing of multiple formulation ingredients, a twin screw extruder (TSE) is preferred because the rotation of the intermeshing screws provides better mixing to produce a homogenous solid dispersion containing finely dispersed API particles or a solid solution of API in polymer. The dissolution or fine dispersion of the API in the molten polymer is achieved by heated extruder barrel and mechanical shear energy provided by the rotating screws as well as barrel wall as the material is conveyed during the HME process.
The scale-up of HME process can be conducted in systematic manner by establishing a link between quality attributes and extrusion parameters by understanding their influence on scale-independent system parameters such as specific energy, residence time distribution, maximum shear rate, temperature profile inside barrel, and degree of fill as shown in Fig. 10 (33,34). The determined link between scale-independent system parameters and drug product quality attributes will remain, regardless of equipment scale or brand. Typically, changes in equipment size will require a determination of the impact of machine parameters such as screw design, die design, free volume, and process parameters such as feed rate, screw speed, barrel temperature on the critical scale-independent system parameters (35). It is important to note that each system parameter is impacted by multiple extrusion parameters with complex interactions; hence, HME scale up studies should be designed carefully. The screw elements of a TSE (used for feeding, conveying, melting, mixing, venting, and pumping) are modular hence customizable. Generally, changes to screw profile cause changes to parameters such as mechanical shear and residence time (36). Feed rate affects the degree to which the extruder screws are filled, which can affect both thermal and mechanical energy input to the formulation.

HME process—scale-independent scale up approach
In the present study, the extrusion process was scaled up from lab scale 9-mm extruder to clinical scale 18-mm extruder. The details of the extruder configuration and evaluated extrusion process conditions are shown in Table I. In general, the lab scale 9-mm extruder was used to conduct feasibility studies to develop various prototypes using only a small amount of API at a batch size of 5–20 g (Fig. 2). While lab scale extruders are useful as a screening tool, they do not scale up to larger-volume extruders because equipment design attributes are fundamentally different. Mid-size extruders in the range of 12–27 mm in diameter that have similar design attributes to larger extruders can be used for process development in preparation for scale up to pilot-size or small commercial-size TSE in the range of 40–70 mm.
The HME process for PVP VA64 containing ASD of compound X was scaled up from 16- to 18-mm extruder using volumetric and power scale up approaches, which have been successfully used by other researchers (37). There were differences in terms of free volume, length/diameter (L/D), and external/internal screw diameter (D o/D i) ratio between 16- and 18-mm extruders; hence, extrusion process conditions were adjusted to achieve similar specific energy range as shown in Table VI (37). Due to lack of geometric similarities between both extruders, the feed rate for the 18-mm extruder was estimated using volumetric scale-up equation as shown in Table VI (37). The estimated feed rate resulted in similar specific energy values when compared to specific energy of 16-mm extruder batch (Table VI). The melt pressure values were higher on 16-mm batch compared to 18-mm batch (Table I), which could be due to differences in die design and opening size between the two extruders. The screw profile used for 16- and 18-mm extruder was not exactly matched. In general, a set of kneading elements and conveying elements (see Fig. 1) were selected for both extruders to achieve adequate mechanical shear energy, which facilitated dissolution of compound X in molten polymer to form an ASD. The differences in screw profile between 16- and 18-mm extruder did not result in any significant difference in mean residence time. The mean residence time of around 60 s was observed at both 16- and 18-mm extruder scale at the selected extrusion process conditions with similar specific energy and the same extrusion temperature (Table VI). The extrudates from 16- and 18-mm extruder were collected for further characterization.
The physico-chemical properties and dissolution performance of PVP VA64 containing milled extrudates of compound X obtained from 9-, 16-, and 18-mm extruder batches were evaluated. An amorphous dispersion was obtained at each scale with similar T g values, which was confirmed by PLM, mDSC, and XRD analysis (Fig. 11a, b). The total impurity value for 9- and 16-mm batch was 0.25% whereas for 18-mm batch, it decreased to 0.14%, which was desirable. The lower level of impurity for 18-mm batch could be due to a higher screw flight depth of 3 mm compared to screw flight depth of 1 mm for 16-mm extruder, which might have resulted in a lower shear rate in screw channels inside the extruder hence lower torque value for 18-mm batch during the extrusion of PVP VA64 containing blend of compound X (Table VI). The dissolution performance was similar from the milled extrudates obtained from 9-, 16- and 18-mm extruder scale by a two-stage non-sink dissolution method (Fig. 11c). Moreover, the extrudate fractions at various time intervals were collected throughout the 18-mm extrusion run and characterized in order to determine the time required to achieve the steady state on 18-mm extruder. The extrudate fractions collected after a 3-min run time were amorphous, had similar T g value of 94°C, similar low impurity value of 0.14%, as well as similar dissolution performance; hence, each collected extrudate fraction after 3 min was combined, which resulted in overall yield of >90% for 18-mm batch.

Solid-state and performance assessment of compound X:PVP VA64 (1:2) ASD from 9- to 18-mm hot melt extruders. a PLM images. b XRPD patterns. c Dissolution profiles
Overall, similar physico-chemical properties and dissolution performance could be achieved for PVP VA64 containing ASD of compound X by successfully scaling up the HME process from lab scale to clinical scale using volumetric scale up approach and scale-independent-specific energy parameter, which resulted in development of robust HME process.
CONCLUSIONS
Systematic development of a bioavailable and stable ASD of compound X by HME process was achieved by using material-sparing methodologies that resulted in selection of suitable polymers for manufacture of ASD using minimal API, developing discriminating non-sink dissolution method which correlated well with the in vivo performance of ASD, conducting thorough evaluation of physical stability of ASD that resulted in selection of stable PVP VA64 containing ASD and suitable packaging configuration. The HME process was successfully scaled up from lab scale to clinical scale using volumetric scale up approach and scale-independent-specific energy parameter, which allowed manufacture of clinical batches and advancement of the amorphous solid dosage form of compound X from early- to late-stage in development with minimal hurdles.
References
Rauwendaal CH. Polymer extrusion. 4th ed. Hanser Gardner Publications. 2012.
Breitenbach J. Melt extrusion can bring new benefits to HIV therapy: the example of Kaletra® tablets. Am J Drug Deliv. 2006;4:61–4.
Crowley MM, Zhang F, Repka MA, Thumma S, Upadhye SB, Battu SK, et al. Pharmaceutical applications of hot melt extrusion: part I. Drug Dev Ind Pharm. 2007;33(9):909–26.
Horspool K, Bhugra C, Agrawal A, Mujumdar S, Telang C, Qiang D. Enabling formulation approaches for poorly soluble compounds in early development. Biopharma Asia. 2012;1:26–35.
Li D, Guo G, Fan R, Liang J, Deng X, Luo F, et al. PLA/F68/Dexamethasone implants prepared by hot-melt extrusion for controlled release of anti-inflammatory drug to implantable medical devices: I. Preparation, characterization and hydrolytic degradation study. Int J Pharm. 2013;441(1–2):365–72.
Madan S, Madan S. Hot melt extrusion and its pharmaceutical applications. Asian J Pharm Sci. 2012;7(2):123–33.
Shah S, Maddineni S, Lu J, Repka MA. Melt extrusion with poorly soluble drugs. Int J Pharm. 2013;453:233–52.
Zhang K, Yu H, Luo Q, Yang S, Lin X, Zhang Y, et al. Increased dissolution and oral absorption of itraconazole/Soluplus extrudate compared with itraconazole nanosuspension. Eur J Pharm Biopharm. 2013;85(3):1285–92.
Repka MA, Majumdar S, Battu SK, Srirangam R, Upadhye SB. Applications of hot melt extrusion for drug delivery. Expert Opin Drug Deliv. 2008;5:1357–76.
Janssens S, Van den Mooter G. Physical chemistry of solid dispersions. J Pharm Pharmacol. 2009;61:1571–86.
Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50:47–60.
Serajuddin ATM. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J Pharm Sci. 1999;88:1058–66.
Weuts I, Van Dycke F, Voorspoels J, De Cort S, Stokbroekx S, Leemans R, et al. Physicochemical properties of the amorphous drug, cast films, and spray dried powders to predict formulation probability of success for solid dispersions: etravirine. J Pharm Sci. 2011;100:260–74.
Baird JA, Taylor LS. Evaluation of amorphous solid dispersion properties using thermal analysis techniques. Adv Drug Deliv Rev. 2012;64:396–421.
Tian W, Caron V, Jones DS, Healy A, Andrews GP. Using Flory-Huggins phase diagrams as a pre-formulation tool for the production of amorphous solid dispersions: a comparison between hot melt extrusion and spray drying. J Pharm Pharmacol. 2013;66:256–74.
Sarode AL, Sandhu H, Shah N, Malick W, Zia H. Hot melt extrusion (HME) for amorphous solid dispersions: predictive tools for processing and impact of drug-polymer interactions on supersaturation. Eur J Pharm Sci. 2013;48:371–84.
Zecevic D, Wagner KG. Rational development of solid dispersions via hot-melt extrusion using screening, material characterization, and numeric simulation tools. J Pharm Sci. 2013;102(7):2297–310.
Abbott S, Hansen CM, Yamamoto H. Hansen solubility parameters in practice. 4th edition version 4.1, e-book. 2013.
Van Krevelen DW. Properties of polymers. Elsevier: Amsterdam. 1990:189–225.
Greenhalgh DJ, William AC, Timmins P, York P. Solubility parameters as predictors of miscibility in solid dispersions. J Pharm Sci. 1999;88:1182–90.
Forster A, Hempenstall J, Tucker I, Rades T. Selection of excipients for melt extrusion with two poorly water soluble drugs by solubility parameter calculation and thermal analysis. Int J Pharm. 2001;226:147–61.
Ghebremeskel AN, Vernavarapu C, Lodaya M. Use of surfactants as plasticizers in preparing solid dispersions of poorly soluble drug: selection of polymer-surfactant combinations using solubility parameters and testing the processability. Int J Pharm. 2007;328(2):119–29.
Dhumal RS, Kelly AL, York P, Coates PD, Paradkar A. Cocrystallization and simultaneous agglomeration using hot melt extrusion. Pharm Res. 2010;27(12):2725–33.
Liu X, Lu M, Guo Z, Huang L, Feng X, Wu C. Improving the chemical stability of amorphous solid dispersion with cocrystal technique by hot melt extrusion. Pharm Res. 2012;29:806–17.
Kolter K, Karl M, Gryczke A. Hot-melt extrusion with BASF pharma polymers. Extrusion compendium. 2nd revision, 2012;104–118.
HELJO V, Nordberg A, Tenho M, Virtanen T, Jouppila K, Salonen J, et al. The effect of water plasticization on the molecular mobility and crystallization tendency of amorphous disaccharides. Pharm Res. 2012;29:2684–97.
Tong P, Zografi G. Effects of water vapor absorption on the physical and chemical stability of amorphous sodium indomethacin. Aaps Pharmsci Tech. 2004;5:9–16.
Khougaz K, Clas SD. Crystallization inhibition in solid dispersions of MK-0591 and poly(vinylpyrrolidone) polymers. J Pharm Sci. 2000;89:1325–34.
Konno H, Taylor LS. Influence of different polymers on the crystallization tendency of molecularly dispersed amorphous felodipine. J Pharm Sci. 2006;95:2692–705.
Matsumoto T, Zografi G. Physical properties of solid molecular dispersions of indomethacin with poly(vinylpyrrolidone) and poly(vinylpyrrolidone-co-vinylacetate) in relation to indomethacin crystallization. Pharm Res. 1999;16:1722–8.
Agrawal A, Dudhedia MS, Patel AD, Raikes MS. Characterization and performance assessment of solid dispersions prepared by hot melt extrusion and spray drying process. Int J Pharm Sci. 2013;457(1):71–81.
Hancock BC, Shamblin SL, Zografi G. Molecular mobility of amorphous pharmaceutical solids below their glass transition temperatures. Pharm Res. 1995;12:799–806.
Horspool K, Agrawal A, Mujumdar S, Bhugra C, Qiang D. Enabling formulation approaches: considerations to advance solid dispersions into late stage development. BioPharma Asia. 2013;2:4–13.
Dreiblatt A. Process design In: Ghebre-Sellassie, I., Martin, C. (eds.), Pharmaceutical Extrusion Technology. Informa Health Care. 2007;133:53–169.
Markarian J. Scale up challenges in hot melt extrusion. Pharm Technol. 2012;36:88–92.
Liu H, Zhu L, Wang P, Zhang X, Gogos CG. Effects of screw configuration on indomethacin dissolution behaviour in Eudragit EPO. Adv Polym Technol. 2011;31(4):331–42.
Thiry J, Krier F, Evrard B. A review of pharmaceutical extrusion: critical process parameters and scaling-up. Int J Pharm. 2015;479:227–40.
Author information
Authors and Affiliations
Corresponding author
Additional information
Guest Editors: Feng Zhang and Michael A. Repka
Rights and permissions
About this article

Cite this article
Agrawal, A.M., Dudhedia, M.S. & Zimny, E. Hot Melt Extrusion: Development of an Amorphous Solid Dispersion for an Insoluble Drug from Mini-scale to Clinical Scale. AAPS PharmSciTech 17, 133–147 (2016). https://doi.org/10.1208/s12249-015-0425-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-015-0425-7