
Abstract
We discuss the design and implementation of StarRate, a modern-Fortran program for the calculation of chemical kinetics coupled to anharmonic vibrational perturbative treatments. The program is written in the F language, a carefully crafted subset of Fortran 95, and is conceived in an object-based programming paradigm, i.e. the set of object-oriented programming features supported by Fortran 90/95. StarRate is made up of three main modules handling the involved molecular species, the elementary reaction steps, and the whole reaction scheme. Input data are accessed through an XML interface based on a cross-code hierarchical data format granting interoperability with popular electronic-structure packages and with the graphical interface of the Virtual Multifrequency Spectrometer developed in our group. Data parsing is performed through versatile Python scripts. Test calculations on the isomerization reaction of C-cyanomethanimine using anharmonic densities of states obtained with a development version of Gaussian are reported together with an account of ongoing developments.
The research leading to these results has received funding from Scuola Normale Superiore through project “DIVE: Development of Immersive approaches for the analysis of chemical bonding through Virtual-reality Environments” (SNS18_B_RAMPINO) and program “Finanziamento a supporto della ricerca di base” (SNS_RB_RAMPINO). The authors are grateful to Dr. Daniele Licari (Scuola Normale Superiore) for fruitful discussions.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Notes
- 1.
Cardelli and Wegner identify using user-defined types for identity and classification without inheritance as object-based programming [6].
- 2.
The partition function Q(T) can as well be computed from the density of states \(\rho (E)\) by a Laplace transform:
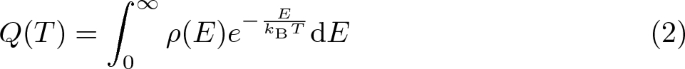
with \(k_\mathrm {B}\) being the Boltzmann constant and T being the absolute temperature.
References
Ram, S.: Dr. Alan Kay on the meaning of Object-Oriented Programming (2003). http://www.purl.org/stefan_ram/pub/doc_kay_oop_en. Accessed 14 Mar 2019
Frisch, M.J., et al.: Gaussian 16 Revision B.01. Gaussian Inc., Wallingford CT (2016)
Cary, J.R., Shasharina, S.G., Cummings, J.C., Reynders, J.V., Hinker, P.J.: Comparison of C++ and Fortran 90 for object-oriented scientific programming. Comput. Phys. Commun. 105(1), 20–36 (1997)
Decyk, V.K., Norton, C.D., Szymanski, B.K.: How to support inheritance and run-time polymorphism in Fortran 90. Comput. Phys. Commun. 115(1), 9–17 (1998)
Gorelik, A.M.: Object-oriented programming in modern Fortran. Program. Comput. Softw. 30(3), 173–179 (2004)
Cardelli, L., Wegner, P.: On understanding types, data abstraction and polymorphism. ACM Comput. Surv. 17(4), 471–522 (1985)
Gray, M.G., Roberts, R.M., Dubois, P.F.: Object-based programming in Fortran 90. Comput. Phys. 11(4), 355–361 (1997)
Kim, Y.H., Lee, I.H., Martin, R.M.: Object-oriented construction of a multigrid electronic-structure code with Fortran 90. Comput. Phys. Commun. 131(1–2), 10–25 (2000)
Jayatilaka, D., Grimwood, D.J.: Tonto: a Fortran based object-oriented system for quantum chemistry and crystallography. In: Sloot, P.M.A., Abramson, D., Bogdanov, A.V., Gorbachev, Y.E., Dongarra, J.J., Zomaya, A.Y. (eds.) ICCS 2003. LNCS, vol. 2660, pp. 142–151. Springer, Heidelberg (2003). https://doi.org/10.1007/3-540-44864-0_15
Zaghi, S.: OFF, open source finite volume fluid dynamics code: a free, high-order solver based on parallel, modular, object-oriented Fortran API. Comput. Phys. Commun. 185(7), 2151–2194 (2014)
Metcalf, M., Reid, J.: The F Programming Language. Oxford University Press Inc., New York (1996)
F Syntax Rules. http://www.fortran.com/F/F_bnf.html. Accessed 14 Mar 2019
Rossi, E., et al.: Code interoperability and standard data formats in quantum chemistry and quantum dynamics: the Q5/D5Cost data model. J. Comput. Chem. 35(8), 611–621 (2014)
Rampino, S., Monari, A., Rossi, E., Evangelisti, S., Laganà, A.: A priori modeling of chemical reactions on computational grid platforms: workflows and data models. Chem. Phys. 398, 192–198 (2012)
Licari, D., Baiardi, A., Biczysko, M., Egidi, F., Latouche, C., Barone, V.: Implementation of a graphical user interface for the Virtual Multifrequency Spectrometer: the VMS-draw tool. J. Comput. Chem. 36(5), 321–334 (2015)
FoX in Fortran Wiki. http://fortranwiki.org/fortran/show/FoX. Accessed 14 Mar 2019
Zaleski, D.P., et al.: Detection of E-cyanomethanimine toward Sagittarius B2(N) in the Green Bank Telescope PRIMOS survey. Astrophys. J. 765(1), L10 (2013)
Melosso, M., et al.: Laboratory measurements and astronomical search for cyanomethanimine. Astron. Astrophys. 609, A121 (2018)
Nandi, S., Bhattacharyya, D., Anoop, A.: Prebiotic chemistry of HCN tetramerization by automated reaction search. Chem. Eur. J. 24(19), 4885–4894 (2018)
Balucani, N.: Elementary reactions and their role in gas-phase prebiotic chemistry. Int. J. Mol. Sci. 10(5), 2304–2335 (2009)
Chakrabarti, S., Chakrabarti, S.K.: Can DNA bases be produced during molecular cloud collapse? Astron. Astrophys. 354, L6–L8 (2000)
Puzzarini, C.: Isomerism of cyanomethanimine: accurate structural, energetic, and spectroscopic characterization. J. Phys. Chem. A 119(47), 11614–11622 (2015)
Vazart, F., Calderini, D., Skouteris, D., Latouche, C., Barone, V.: Reassessment of the thermodynamic, kinetic, and spectroscopic features of cyanomethanimine derivatives: a full anharmonic perturbative treatment. J. Chem. Theor. Comput. 11(3), 1165–1171 (2015)
Rampino, S., Faginas Lago, N., Laganà, A., Huarte-Larrañaga, F.: An extension of the grid empowered molecular simulator to quantum reactive scattering. J. Comput. Chem. 33(6), 708–714 (2012)
Rampino, S., Skouteris, D., Laganà, A.: Microscopic branching processes: the O + O\(_2\) reaction and its relaxed potential representations. Int. J. Quantum Chem. 110(2), 358–367 (2010)
Rampino, S., Skouteris, D., Laganà, A.: The O + O\(_2\) reaction: quantum detailed probabilities and thermal rate coefficients. Theor. Chem. Acc. 123(3/4), 249–256 (2009)
Laganà, A., Faginas Lago, N., Rampino, S., Huarte Larrañaga, F., García, E.: Thermal rate coefficients in collinear versus bent transition state reactions: the N + N\(_2\) case study. Physica Scripta 78(5), 058116 (2008)
Rampino, S., Pastore, M., Garcia, E., Pacifici, L., Laganà, A.: On the temperature dependence of the rate coefficient of formation of C\(_2^+\) from C + CH\(^+\). Monthly Not. Roy. Astron. Soc. 460(3), 2368–2375 (2016)
Pacifici, L., Pastore, M., Garcia, E., Laganà, A., Rampino, S.: A dynamics investigation of the C + CH\(^+\)\(\rightarrow \) C\(_2^+\) + H reaction on an ab initio bond-order like potential. J. Phys. Chem. A 120(27), 5125–5135 (2016)
Rampino, S., Suleimanov, Y.V.: Thermal rate coefficients for the astrochemical process C + CH\(^+\)\(\rightarrow \) C\(2^+\) + H by ring polymer molecular dynamics. J. Phys. Chem. A 120(50), 9887–9893 (2016)
Rice, O.K., Ramsperger, H.C.: Theories of unimolecular gas reactions at low pressures. J. Am. Chem. Soc. 49(7), 1617–1629 (1927)
Kassel, L.S.: Studies in homogeneous gas reactions. I. J. Phys. Chem. 32(2), 225–242 (1927)
Marcus, R.A.: Unimolecular dissociations and free radical recombination reactions. J. Chem. Phys. 20(3), 359–364 (1952)
Brouard, M.: Reaction Dynamics. Oxford Chemistry Primers. OUP Oxford (1998)
Green, N.J.B.: Chapter 1 - introduction. In: Green, N.J., (ed.) Unimolecular Kinetics. Volume 39 of Comprehensive Chemical Kinetics, pp. 1–53. Elsevier (2003)
Barone, V.: Anharmonic vibrational properties by a fully automated second-order perturbative approach. J. Chem. Phys. 122(1), 014108 (2005)
Bloino, J., Biczysko, M., Barone, V.: General perturbative approach for spectroscopy, thermodynamics, and kinetics: methodological background and benchmark studies. J. Chem. Theor. Comput. 8(3), 1015–1036 (2012)
Stein, S.E., Rabinovitch, B.S.: Accurate evaluation of internal energy level sums and densities including anharmonic oscillators and hindered rotors. J. Chem. Phys. 58(6), 2438–2445 (1973)
Beyer, T., Swinehart, D.F.: Algorithm 448: number of multiply-restricted partitions. Commun. ACM 16(6), 379 (1973)
Wang, F., Landau, D.P.: Efficient, multiple-range random walk algorithm to calculate the density of states. Phys. Rev. Lett. 86(10), 2050–2053 (2001)
Zhou, C., Bhatt, R.N.: Understanding and improving the Wang-Landau algorithm. Phys. Rev. E 72(2), 025701 (2005)
Basire, M., Parneix, P., Calvo, F.: Quantum anharmonic densities of states using the Wang-Landau method. J. Chem. Phys. 129(8), 081101 (2008)
Nguyen, T.L., Barker, J.R.: Sums and densities of fully coupled anharmonic vibrational states: a comparison of three practical methods. J. Phys. Chem. A 114(10), 3718–3730 (2010)
Frisch, M.J., et al.: Gaussian development version, revision i.13. Gaussian Inc., Wallingford CT (2018)
Lee, C., Yang, W., Parr, R.G.: Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988)
Becke, A.D.: Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98(7), 5648–5652 (1993)
Grimme, S., Ehrlich, S., Goerigk, L.: Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32(7), 1456–1465 (2011)
Double and triple-\(\zeta \) basis sets of SNS families are available for download. http://smart.sns.it. Accessed 14 Mar 2019
Bloino, J., Barone, V.: A second-order perturbation theory route to vibrational averages and transition properties of molecules: general formulation and application to infrared and vibrational circular dichroism spectroscopies. J. Chem. Phys. 136(12), 124108 (2012)
Rampino, S.: Configuration-space sampling in potential energy surface fitting: a space-reduced bond-order grid approach. J. Phys. Chem. A 120(27), 4683–4692 (2016)
Salvadori, A., Fusè, M., Mancini, G., Rampino, S., Barone, V.: Diving into chemical bonding: an immersive analysis of the electron charge rearrangement through virtual reality. J. Comput. Chem. 39(31), 2607–2617 (2018)
Bistoni, G., Rampino, S., Tarantelli, F., Belpassi, L.: Charge-displacement analysis via natural orbitals for chemical valence: charge transfer effects in coordination chemistry. J. Chem. Phys. 142(8), 084112 (2015)
Fusè, M., Rimoldi, I., Cesarotti, E., Rampino, S., Barone, V.: On the relation between carbonyl stretching frequencies and the donor power of chelating diphosphines in nickel dicarbonyl complexes. Phys. Chem. Chem. Phys. 19, 9028–9038 (2017)
Fusè, M., Rimoldi, I., Facchetti, G., Rampino, S., Barone, V.: Exploiting coordination geometry to selectively predict the \(\sigma \)-donor and \(\pi \)-acceptor abilities of ligands: a back-and-forth journey between electronic properties and spectroscopy. Chem. Commun. 54, 2397–2400 (2018)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this paper

Cite this paper
Nandi, S., Calderini, D., Bloino, J., Rampino, S., Barone, V. (2019). A Modern-Fortran Program for Chemical Kinetics on Top of Anharmonic Vibrational Calculations. In: Misra, S., et al. Computational Science and Its Applications – ICCSA 2019. ICCSA 2019. Lecture Notes in Computer Science(), vol 11624. Springer, Cham. https://doi.org/10.1007/978-3-030-24311-1_29
Download citation
DOI: https://doi.org/10.1007/978-3-030-24311-1_29
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-24310-4
Online ISBN: 978-3-030-24311-1
eBook Packages: Computer ScienceComputer Science (R0)