
Abstract
Severe acute respiratory syndrome (SARS) is an acute infectious disease with significant mortality. A novel coronavirus (SARS-CoV) has been shown to be the causative agent of SARS. The typical clinical feature associated with SARS is diffuse alveolar damage in lung, and lung fibrosis is evident in patients who died from this disease. The mechanisms by which SARS-CoV infection causes lung fibrosis are not fully understood, but transforming growth factor-β (TGF-β) and angiotensin-converting enzyme 2 (ACE2)-mediated lung fibrosis are among the most documented ones. The activation of the TGF-β/Smad pathway is critical to lung fibrosis. SARS-CoV infection not only enhances the expression of TGF-β, but also facilitates its signaling activity. The SARS-CoV receptor ACE2 is a negative regulator of lung fibrosis, and SARS-CoV infection decreases ACE2 expression. Therefore, SARS-CoV infection may lead to lung fibrosis through multiple signaling pathways and TGF-β activation is one of the major contributors.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Severe Acute Respiratory Syndrome
- Connective Tissue Growth Factor
- Alveolar Epithelial Cell
- Lung Fibrosis
- Severe Acute Respiratory Syndrome
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Severe acute respiratory syndrome (SARS) is an acute infectious disease with significant morbidity and mortality (Wang and Chang 2004). An intense, cooperative worldwide effort led to the identification of the etiological agent as a novel SARS coronavirus (SARS-CoV) (Guan et al. 2003; Peiris et al. 2003; Rota et al. 2003) and the subsequent complete sequencing of the viral genome. SARS-CoV belongs to a family of large, positive, single-stranded RNA viruses. The SARS-CoV genome is 29.7 kb in length, and encodes 14 putative open reading frames generating 28 potential proteins, the functions of many of which are not known (Marra et al. 2003). Sequence comparison with other known coronaviruses revealed a similar organization of SARS-CoV genes to typical coronaviruses (Chen et al. 2006).
Coronaviruses are known to cause up to 30% of common colds in humans but their infection leads only to lower respiratory tract diseases in livestock and poultry. However, SARS-CoV infection results in severe and even fatal lung disease in humans. At 1–2 weeks after the onset of the disease, the majority of patients can recover but one-third of patients develop severe symptoms. Many patients in the latter group deteriorate into acute respiratory distress syndrome (ARDS) with high mortality. During the convalescence, patients gradually shed virus but this process may take a long time. However, no chronic infection by SARS-CoV has been documented in humans. The virus can be found in respiratory tract secretions, lung tissue, serum, and stool (Tse et al. 2004).
According to the data from autopsy of fatal cases, the major pathological characteristic in SARS patients is diffuse alveolar damage (DAD) (Chan et al. 2003). The mechanism of DAD is believed to be endothelial and alveolar epithelial injury due to both direct viral effects and other indirect factors (Nicholls et al. 2003). During the early phase of SARS development (7–10 days), the lungs demonstrate the features of DAD including extensive edema, hyaline membrane formation, fluid and cellular exudation, collapse of alveoli, and desquamation of alveolar epithelial cells. At the same time, fibrous tissue could be detected in alveolar spaces. This phase is referred as the acute stage. In the medium phase of SARS development (10–14 days), the lungs display fibrous organization including interstitial and airspace fibrosis, reparative fibroblastic proliferation and type II pneumocytic hyperplasia. The fibrous organization becomes more extensive with time. This phase is referred as the proliferative organizing stage. In the late phase of SARS (2–3 weeks), referred as the fibrotic stage, the lungs exhibit dense septal and alveolar fibrosis. The time duration is counted from onset of the symptoms, and the pathological process does not always evolve through all the three stages, but may cease or recover at any phase (Chan et al. 2003; Cheung et al. 2004; Gu and Korteweg 2007; Guo et al. 2008; Ketai et al. 2006; Ng et al. 2006; Nicholls et al. 2003; Tse et al. 2004).
The extent of lung fibrosis is positively correlated with the duration of the SARS disease. Clinical data showed that fibrous organization is more common in patients in the late phase than in patients in the early or medium phases. In contrast, DAD more likely happens in patients in the early or medium phases than in patients in the late phase. Thus lung fibrosis can also display without DAD (Hwang et al. 2005). Importantly, lung fibrosis was even observed in SARS patients who had recovered and been discharged from hospital. Furthermore, the prevalence rate of lung fibrosis in cured SARS patients at 9 months after discharge is about 21% (42/200). The traditional therapy for lung fibrosis (glucocorticoid and hormone treatment) has no effect on the SARS-induced lung fibrosis regardless of the dosage, method, or length of drug usage. Interestingly, individuals who had lung fibrosis showed some spontaneous recovery (Xie et al. 2005).
2 SARS-CoV-Mediated Lung Fibrosis
Lung fibrosis is a pathological consequence of acute and chronic interstitial lung diseases. Lung fibrosis is characterized by failed alveolar re-epithelialization, fibroblast persistence, and excessive deposition of collagen and other extracellular matrix (ECM) components as well as destruction of the normal lung architecture (Sime and O'Reilly 2001). Progression of lung fibrosis results in the widening of interstitial matrix, eventual compression and destruction of normal lung parenchyma, and resultant damage to the capillaries leading to ventilatory insufficiency (Razzaque and Taguchi 2003). The etiology of lung fibrosis is uncertain: smoking, viral infection, drug exposure, and genetic predisposition may contribute to the fibrotic process. Chronic inflammation was regarded as the main reason for lung fibrosis and it can result in epithelial injury and fibroblast activation. However, recent studies have suggested that alveolar epithelial injury and formation of active myofibroblast foci are the main reasons for most lung fibrotic processes (Chapman 2004; Gharaee-Kermani et al. 2007; Kalluri and Neilson 2003; Razzaque and Taguchi 2003; Scotton and Chambers 2007).
Once damage takes place in lung tissue, a set of growth factors and cytokines, including monocyte chemoattractant protein-1 (MCP-1), transforming growth factor-β1 (TGF-β1), tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6), are overexpressed and released by the cells. Type II alveolar epithelial cells are one of the major sources of these fibrogenic factors. These factors in turn stimulate hyperproliferation of type II alveolar epithelial cells, recruit fibroblasts to the fibrotic loci, and induce transdifferentiation/activation of fibroblasts into myofibroblasts. The myofibroblasts are responsible for the excessive accumulation of ECM in the basement membranes and interstitial tissues, which finally leads to the loss of alveolar function, especially gas exchange between alveoli and capillaries (Razzaque and Taguchi 2003).
The excessive accumulation of ECM may result from increased synthesis or decreased degradation of ECM or both. The expression and deposition of collagen are the major events of ECM accumulation. Various types of collagens, such as type I, type III, and type VI, have been found in fibrotic lesions of the lungs. Many factors are involved in the ECM degradation, amongst which the plasminogen activator/plasmin system plays an important role. The activity of plasminogen is regulated by a physiologic inhibitor, plasminogen activator inhibitor 1 (PAI-1). In fibrotic lungs of patients, PAI-1 expression is increased. The deletion of the PAI-1 gene can reduce the susceptibility of mouse to lung fibrosis induced by various stimuli whereas PAI-1 protein overexpression enhances it, indicating that PAI-1 is critical in the development of lung fibrosis (Izuhara et al. 2008). In addition to plasminogen, other proteolytic enzymes including matrix metalloproteinases (MMP) and a disintegrin and metalloproteinase domain (ADAM) also contribute to ECM degradation. The tissue inhibitors of metalloproteinases (TIMP) accelerate EMC deposition by neutralizing MMP activities (Gill and Parks 2008). In fibrotic lungs of patients, a wider distribution of TIMP compared with MMP has been described, suggesting that in lung fibrosis the total ECM degradation is reduced (Gill and Parks 2008; Izuhara et al. 2008; Liu 2008; Razzaque and Taguchi 2003).
3 TGF-β and SARS-Induced Lung Fibrosis
3.1 TGF-β Signal Transduction
TGF-β is a secreted cytokine which belongs to a superfamily of structure-related growth factors including TGF-β, activin, bone morphogenetic proteins (BMPs), and others. TGF-β signaling is initiated via its binding to the transmembrane receptors (Heldin et al. 1997; Hu et al. 2006; Massague and Chen 2000; Shi and Massague 2003). Ligand binding results in the formation of heterocomplexes between type I and type II receptors, in which the type II receptor phosphorylates and activates the type I receptor. The activated type I receptor then phoshorylates downstream signaling mediators – R-Smad proteins – which form a heteromeric complex with common Smad–Smad4 and are accumulated in the nucleus to orchestrate the expression of target genes in collaboration with other DNA-binding factors or transcription cofactors. In addition to the canonical Smad pathway, TGF-β has been reported to activate mitogen-activated protein kinases (ERK, p38 and JNK), phosphoinositide-3 kinase (PI3K)/Akt, p21-activated kinase (PAK), and others (Derynck and Akhurst 2007; Moustakas and Heldin 2005).
The TGF-β superfamily members play multifunctional regulatory roles in cell growth, differentiation, death, and migration and matrix remodeling (Derynck and Miyazono 2008; Hu et al. 2006; Massague and Chen 2000). Dysregulation of TGF-β signaling has been associated with a variety of human diseases, such as cancer, tissue fibrosis, and cardiovascular disorders (Derynck and Miyazono 2008; Massague and Chen 2000; ten Dijke and Arthur 2007).
3.2 TGF-β and Lung Fibrosis
Transgenic studies on an animal model reveal the importance of TGF-β activation in the process of lung fibrosis. Using replication-deficient adenovirus vectors to transfer the cDNA of TGF-β1 to rat lung, the effect of TGF-β1 protein in the respiratory tract was directly tested. Transient overexpression of active TGF-β1 resulted in severe interstitial and pleural fibrosis (Sime et al. 1997). Soluble TGF-β type II receptor consisting of the extracellular domain, which can bind to TGF-β and function as a TGF-β antagonist, has been shown to inhibit lung fibrosis in an animal model (Lee et al. 2001).
TGF-β activation is the hallmark event in lung fibrotic processes. In fibrotic lung tissue, the levels of TGF-β mRNA as well as TGF-β protein are increased (Rube et al. 2000). TGF-β stimulates the production of ECM proteins, enhances the secretion of protease inhibitors (PAI-1 and TIMP), and reduces the secretion of proteases, thus leading to deposition of ECM proteins (Liu and Matsuura 2005). Blocking the activity of TGF-β or disrupting TGF-β signaling cascades in animal models could inhibit matrix production and prevent the fibrotic process (Liu et al. 2005; Rube et al. 2000; Willis et al. 2005).
TGF-β1 can also promote lung fibrosis through induction of myofibroblast expansion. As mentioned above, myofibroblasts are the key effector cells in lung fibrogenesis. Myofibroblasts localize to fibrotic foci and other sites of active fibrosis, and contribute to most ECM production. TGF-β can directly promote resident lung fibroblasts to differentiate into myofibroblasts. Furthermore, TGF-β induces the epithelial cells to undergo transition to form fibroblasts and hence myofibroblasts, a process termed epithelial–mesenchymal transition (EMT) (Derynck and Akhurst 2007). It has been shown that alveolar epithelial cells can undergo EMT in response to TGF-β both in vitro and in animal models (Kim et al. 2006; Willis et al. 2005). In biopsy samples, both epithelial and mesenchymal markers were found to be coexpressed in a cell, suggesting the existence of the EMT state in fibrotic lungs (Burkett et al. 2007; Kim et al. 2006; Willis et al. 2005; Wu et al. 2007). Thus, TGF-β can induce lung fibrosis through multiple mechanisms.
3.3 The Role of TGF-β in SARS-Induced Lung Fibrosis
Consistent with the promoting effect of TGF-β in tissue fibrosis, viral infection (such as HIV, HCV, and HBV infection) has been reported to upregulate TGF-β expression in various tissues (Lin et al. 2007; Ray et al. 2003). In the case of SARS-CoV infection, the serum level of TGF-β1 was elevated during the early phase of SARS (Pang et al. 2003). A high level of TGF-β was also observed in SARS-CoV-infected lung cells (including alveolar epithelial cells, bronchial epithelial cells, and monocytes/macrophages), but not in uninfected lung cells (Baas et al. 2006; He et al. 2006). The virus-induced high level of serum and in situ TGF-β ligand leads to hyperactivation of the TGF-β pathway, thus promoting the lung fibrosis. However, the mechanism by which SARS-CoV infection modulates the tissue level of TGF-β is unknown.
Importantly, SARS-CoV not only modulates TGF-β expression, but also directly regulates the signal transduction process of the TGF-β pathway through viral nucleocapsid (N) protein. The SARS-CoV N protein is a 46 kDa viral RNA-binding protein and shares little homology with the counterparts of other known coronaviruses (He et al. 2004). Our recent study demonstrated that N protein can associate with Smad3 and interferes with the formation of the complex between Smad3 and Smad4 (Zhao et al. 2008). However, N protein has no effect either on TGF-β-stimulated phosphorylation or on the nuclear accumulation of Smad3. Furthermore, it could promote the interaction between Smad3 and the transcriptional coactivator p300 to activate downstream genes. In lung epithelial and fibroblast cells, overexpressing N protein enhances the TGF-β-induced expression of PAI-1 and collagen I, and this enhancement is independent of Smad4. Interestingly, N protein attenuates Smad4-dependent apoptosis, which is in agreement with the fact that N protein was not found in the apoptotic cells (Zhao et al. 2008). Thus, SARS-CoV can promote the lung fibrosis process through its N protein.
4 ACE2/Angiotensin II and SARS-Induced Lung Fibrosis
The renin–angiotensin II system is an important regulator of blood pressure homeostasis. Angiotensin-converting enzyme (ACE) plays a key role in generating angiotensin II (ANG-II) from ANG-I which has no biological function. ACE2 is a recently identified homologue of ACE, which decreases ANG-II level by cleaving ANG-II. Therefore, ACE2 plays an opposite role to ACE in ANG-II generation. In respiratory tract, ACE2 is expressed on the surface of tracheobronchial and alveolar epithelium (Kuba et al. 2006a).
ACE2 has been reported to have a protective role in lung fibrosis. In lung biopsy specimens from patients with lung fibrosis, the ACE2 mRNA and enzyme activity are decreased by 92% (p <0.01) and 74% (p <0.05), respectively (Li et al. 2008). In a mouse model of bleomycin-induced lung fibrosis, intratracheal administration of ACE2-specific small interfering RNAs increased the accumulation of lung collagen, while recombinant human ACE2 decreased the accumulation of lung collagen (Imai et al. 2008; Kuba et al. 2006a, 2006b).
It is suggested that ACE2 protects against lung fibrosis through negative regulation of the local ANG-II level. In addition to its role in vasoconstriction regulation, ANG-II also contributes to many fibrotic diseases including lung, renal, hepatic, and cardiac fibrosis (Watanabe et al. 2005). ANG-II is mainly produced by fibroblasts, as well as activated macrophages. ANG-II signals via two receptors AGTR1 and AGTR2. ANG-II could stimulate the production and secretion of TGF-β cytokine in lung tissue, and this process may be mediated by AGTR1 (Molteni et al. 2007). As TGF-β itself can also regulate the level of ANG-II, it is believed that an “autocrine loop” between ANG-II and TGF-β exists in lung tissue. Therefore, both ACE inhibitor and AGTR antagonist can block lung fibrosis in experimental animals (Uhal et al. 2007). In a mouse model of bleomycin-induced lung fibrosis, the antagonist of angiotensin II receptor reduces the lung fibrosis score. This effect is accompanied with a reduction of the TGF-β level (Otsuka et al. 2004; Waseda et al. 2008).
In addition to regulating the TGF-β ligand level, ANG-II also increases the intracellular Smad2 and Smad4 protein levels and facilitates nuclear translocation of phosphorylated Smad2 (Hao et al. 2000). It has been recently reported that ANG-II activates Smad signaling in a TGF-β-independent and MAPK-dependent manner (Su et al. 2006). ANG-II also induces the EMT process. This effect can be blocked by a TGF-β antagonist Smad7 (Rodriguez-Vita et al. 2004). Altogether, ANG-II enhances TGF-β/Smad signaling and the interplay between ANG-II and TGF-β may promote lung fibrosis.
In addition to enhancing TGF-β expression, ANG-II has been reported to upregulate connective tissue growth factor (CTGF) (Rodriguez-Vita et al. 2005). CTGF may promote deposition of ECM and lung fibrosis via the MEK/Erk pathway (Ponticos et al. 2003). ANG-II can also directly upregulate the level of α-SMA, a key protein in pathogenesis of lung fibrosis (Molteni et al. 2007), as well as production of ECM (Rodriguez-Vita et al. 2005).
Several studies have verified ACE2 as an essential receptor for SARS-CoV infection. The spike protein of SARS-CoV could bind to ACE2 on the cell surface, and this binding promotes viral entry into the cell and induces cell–cell fusion into multinucleated cells (syncytia formation) (Li et al. 2003). ACE2 proteins are expressed by alveolar epithelial cells, the primary targets of SARS-CoV in lung. Thus, ACE2 contributes to SARS disease both as protector of lung fibrosis and as the cellular receptor for viral infection (Imai et al. 2005). Interestingly, SARS-CoV infection and the spike protein of SARS-CoV reduce ACE2 expression level during viral infection (Kuba et al. 2005). As discussed above, the decreased ACE2 expression results in an increased ANG-II level and leads to lung fibrosis and lung failure. Accordingly, the in-vivo administration of the spike protein did not affect the severity of lung failure in Ace2 knockout mice (Kuba et al. 2005). Furthermore, administration of spike protein to mice led to a significant increase in ANG-II level in the lung tissue. Inhibition of ANG-II receptor can rescue the effect of the spike protein (Imai et al. 2007; Kuba et al. 2005). Altogether, infection with SARS-CoV results in ACE2 downregulation through the binding of SARS-CoV spike protein to ACE2, and this spike protein-mediated ACE2 downregulation is responsible for the pathogenesis of lung fibrosis by upregulating ANG-II and activating TGF-β signaling.
5 Other Mechanisms of SARS-Mediated Lung Fibrosis
Besides TGF-β and ACE2, other mechanisms may contribute to SARS-CoV-mediated lung fibrosis. MCP-1 is a chemokine that promote lung fibrosis. In patients with lung fibrosis, the MCP-1 level is usually upregulated (Emad and Emad 2007; Wynn 2008). SARS-CoV infection upregulates the MCP-1 level in patients. The elevation of the MCP-1 level happens at least 2 weeks after disease onset. Corticosteroid treatment reduces MCP-1 concentrations at 5–8 days after treatment (Wong et al. 2004). MAPK p38 is a signal transducer that responds to extracellular stimulation including viral infection, and in turn regulates cell differentiation and ECM production. Hyperactivation of p38 was detected in patients of lung fibrosis, and phosphorylation of p38 is responsible for pulmonary myofibroblast activation and α-SMA expression (Hu et al. 2006; Yoshida et al. 2002). SARS-CoV infection has been reported to induce p38 phosphorylation in lung epithelial and other cells, which lead to actin reorganization (Mizutani et al. 2006; Surjit et al. 2004). Thus, p38 may be involved in SARS-CoV-mediated lung fibrosis.
6 Conclusion
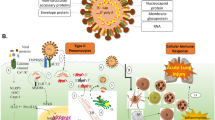
Fibrosis usually brings irreversible damage to the lung. Lung fibrosis is widely observed in patients who died from SARS. However, the mechanisms by which SARS-CoV infection leads to lung fibrosis remain poorly understood. We have discussed two important mechanisms for SARS-mediated fibrosis: activation of TGF-β signaling and degradation of ACE2 (Fig. 15.1). A major challenge will be how to use what we have learned to prevent SARS-CoV-evoked lung fibrosis.


The possible molecular mechanisms of SARS-CoV-mediated lung fibrosis. SARS-CoV infection upregulates TGF-β expression while downregulating the level of the SARS-CoV receptor ACE2. The decreased ACE2 expression leads to a high ANG-II level, which further enhances the TGF-β level. TGF-β activates Smad proteins via receptor-mediated phosphorylation and stimulates Smad-dependent gene transcription, which is facilitated by N protein of SARS-CoV and ANG-II. Both the TGF-β/Smad and the ACE2/ANG-II/CTGF pathways contribute to myofibroblast activation and ECM accumulation, collectively leading to the final lung fibrosis
References
Baas T, Taubenberger JK, Chong PY, Chui P, Katze MG (2006) SARS-CoV virus-host interactions and comparative etiologies of acute respiratory distress syndrome as determined by transcriptional and cytokine profiling of formalin-fixed paraffin-embedded tissues. J Interferon Cytokine Res 26:309–317
Burkett PR, Noth I, Husain AN (2007) Evidence of epithelial/mesenchymal transition in idiopathic pulmonary fibrosis. Lab Invest 87:319a
Chan KS, Zheng JP, Mok YW, Li YM, Liu YN, Chu CM, Ip MS (2003) SARS: prognosis, outcome and sequelae. Respirology 8:S36–S40
Chapman HA (2004) Disorders of lung matrix remodeling. J Clin Invest 113:148–157
Chen SA, Luo HB, Chen LL, Chen J, Shen JH, Zhu WL, Chen KX, Shen X, Jiang HL (2006) An overall picture of SARS coronavirus (SARS-CoV) genome-encoded major proteins: Structures, functions and drug development. Curr Pharm Des 12:4539–4553
Cheung OY, Chan JWM, Ng CK, Koo CK (2004) The spectrum of pathological changes in severe acute respiratory syndrome (SARS). Histopathology 45:119–124
Derynck R, Akhurst RJ (2007) Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat Cell Biol 9:1000–1004
Derynck R, Miyazono K (2008) The TGF-beta family. Cold Spring Harbor Laboratory, Cold Spring Harbor
Emad A, Emad V (2007) Elevated levels of MCP-1, MIP-alpha and MIP-1 beta in the bronchoalveolar lavage (BAL) fluid of patients with mustard gas-induced pulmonary fibrosis. Toxicology 240:60–69
Gharaee-Kermani M, Gyetko MR, Hu B, Phan SH (2007) New insights into the pathogenesis and treatment of idiopathic pulmonary fibrosis: a potential role for stem cells in the lung parenchyma and implications for therapy. Pharm Res 24:819–841
Gill SE, Parks WC (2008) Metalloproteinases and their inhibitors: regulators of wound healing. Int J Biochem Cell Biol 40:1334–1347
Gu J, Korteweg C (2007) Pathology and pathogenesis of severe acute respiratory syndrome. Am J Pathol 170:1136–1147
Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Luo SW, Li PH, Zhang LJ, Guan YJ et al (2003) Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302:276–278
Guo Y, Korteweg C, McNutt MA, Gu J (2008) Pathogenetic mechanisms of severe acute respiratory syndrome. Virus Res 133:4–12
Hao JM, Wang BQ, Jones SC, Jassal DS, Dixon IMC (2000) Interaction between angiotensin II and Smad proteins in fibroblasts in failing heart and in vitro. Am J Physiol Heart Circ Physiol 279:H3020–H3030
He RT, Dobie F, Ballantine M, Leeson A, Li Y, Bastien N, Cutts T, Andonov A, Cao JX, Booth TF et al (2004) Analysis of multimerization of the SARS coronavirus nucleocapsid protein. Biochem Biophys Res Commun 316:476–483
He L, Ding Y, Zhang Q, Che X, He Y, Shen H, Wang H, Li Z, Zhao L, Geng J et al (2006) Expression of elevated levels of pro-inflammatory cytokines in SARS-CoV-infected ACE2(+) cells in SARS patients: relation to the acute lung injury and pathogenesis of SARS. J Pathol 210:288–297
Heldin CH, Miyazono K, ten Dijke P (1997) TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 390:465–471
Hu YB, Peng JW, Feng DY, Chu L, Li XA, Jin ZY, Lin Z, Zeng QF (2006) Role of extracellular signal-regulated kinase, p38 kinase, and activator protein-1 in transforming growth factor-beta 1-induced alpha smooth muscle actin expression in human fetal lung fibroblasts in vitro. Lung 184:33–42
Hwang DM, Chamberlain DW, Poutanen SM, Low DE, Asa SL, Butany J (2005) Pulmonary pathology of severe acute respiratory syndrome in Toronto. Mod Pathol 18:1–10
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H et al (2005) Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436:112–116
Imai Y, Kuba K, Penninger JM (2007) Angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Cell Mol Life Sci 64:2006–2012
Imai Y, Kuba K, Penninger JM (2008) The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp Physiol 93:543–548
Izuhara Y, Takahashi S, Nangaku M, Takizawa SY, Ishida H, Kurokawa K, de Strihou CV, Hirayama N, Miyata T (2008) Inhibition of plasminogen activator inhibitor-1: its mechanism and effectiveness on coagulation and fibrosis. Arteriosclerosis Thrombosis Vasc Biol 28:672–677
Kalluri R, Neilson EG (2003) Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest 112:1776–1784
Ketai L, Paul NS, Wong KTT (2006) Radiology of severe acute respiratory syndrome (SARS) – The emerging pothologic-radiologic correlates of an emerging disease. J Thoracic Imag 21:276–283
Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA (2006) Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A 103:13180–13185
Kuba K, Imai Y, Rao SA, Gao H, Guo F, Guan B, Huan Y, Yang P, Zhang YL, Deng W et al (2005) A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med 11:875–879
Kuba K, Imai Y, Penninger JM (2006a) Angiotensin-converting enzyme 2 in lung diseases. Curr Opin Pharmacol 6:271–276
Kuba K, Imai Y, Rao S, Jiang CY, Penninger JM (2006b) Lessons from SARS: control of acute lung failure by the SARS receptor ACE2. J Mol Med 84:814–820
Lee CG, Homer R, Zhou Z, Lanone Z, Wang XM, Koteliansky V, Shipley JM, Gotwals P, Noble P, Chen QS et al (2001) Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med 194:809–821
Liu F, Matsuura I (2005) Inhibition of Smad antiproliferative function by CDK phosphorylation.Cell Cycle 4:63–66
Li WH, Moore MJ, Vasilieva N, Sui JH, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC et al (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454
Li XP, Molina-Molina M, Abdul-Hafez A, Uhal V, Xaubet A, Uhal BD (2008) Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am J Physiol Lung Cell Mol Physiol 295:L178–L185
Lin WY, Weinberg E, Kim KA, Peng LF, Kim SS, Brockman M, Lopez-Marra H, De Sa Borges CB, Hyppolite G, Shao RX, Chung RT (2007) Hiv and GP120 enhance HCV replication and upregulate TGF-beta 1. Hepatology 46:447a
Liu RM (2008) Oxidative stress, plasminogen activator inhibitor 1, and lung fibrosis. Antioxidants Redox Signal 10:303–319
Liu RM, Vayalil P, Wang SQ, Ballinger C, Gauldie J, Postlethwait E (2005) TGF-beta1 induces concomitant lung fibrosis, decreased GSH and ascorbate concentrations, and increased PAI-1 gene expression. Free Radical Biol Med 39:S37
Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YS, Khattra J, Asano JK, Barber SA, Chan SY et al (2003) The Genome sequence of the SARS-associated coronavirus. Science 300:1399–1404
Massague J, Chen YG (2000) Controlling TGF-beta signaling. Genes Dev 14:627–644
Mizutani T, Fukushi S, Ishii K, Sasaki Y, Kenri T, Saijo M, Kanaji Y, Shirota K, Kurane I, Morikawa S (2006) Mechanisms of establishment of persistent SARS-CoV-infected cells. Biochem Biophys Res Commun 347:261–265
Molteni A, Wolfe LF, Ward WF, Ts'ao CH, Molteni LB, Veno P, Fish BL, Taylor JM, Quintanilla N, Herndon B, Moulder JE (2007) Effect of an angiotensin II receptor blocker and two angiotensin converting enzyme inhibitors on transforming growth factor-beta (TGF-beta) and alpha-actomyosin (alpha SMA), important mediators of radiation-induced pneumopathy and lung fibrosis. Curr Pharm Des 13:1307–1316
Moustakas A, Heldin CH (2005) Non-Smad TGF-beta signals. J Cell Sci 118:3573–3584
Ng WF, To KF, Lam WWL, Ng TK, Lee KC (2006) The comparative pathology of severe acute respiratory syndrome and avian influenza A subtype H5N1 – a review. Hum Pathol 37:381–390
Nicholls J, Dong XP, Jiang G, Peiris M (2003) SARS: clinical virology and pathogenesis. Respirology 8:S6–S8
Otsuka M, Takahashi H, Shiratori M, Chiba H, Abe S (2004) Reduction of bleomycin induced lung fibrosis by candesartan cilexetil, an angiotension II type 1 receptor antagonist. Thorax 59:31–37
Pang BS, Wang Z, Zhang LM, Tong ZH, Xu LL, Huang XX, Guo WJ, Zhu M, Wang C, Li XW et al (2003) Dynamic changes in blood cytokine levels as clinical indicators in severe acute respiratory syndrome. Chin Med J 116:1283–1287
Peiris JSM, Lai ST, Poon LLM, Guan Y, Yam LYC, Lim W, Nicholls J, Yee WKS, Yan WW, Cheung MT et al (2003) Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319–1325
Ponticos M, Holmes AM, Rajkumar V, Wen XS, Leask A, Abraham DJ, Black CM (2003) Connective tissue growth factor (CTGF) mediates extracellular matrix deposition in bleomycin-induced lung fibrosis via the MEK/ERK signaling pathways. Arthritis Rheum 48:S156
Ray S, Broor SL, Vaishnav Y, Sarkar C, Girish R, Dar L, Seth P, Broor S (2003) Transforming growth factor beta in hepatitis C virus infection: in vivo and in vitro findings. J Gastroenterol Hepatol 18:393–403
Razzaque MS, Taguchi T (2003) Pulmonary fibrosis: Cellular and molecular events. Pathol Int 53:133–145
Rodriguez-Vita J, Sanchez-Lopez E, Esteban V, Ruperez M, Lopez A, Egido J, Ruiz-Ortega M (2004) Angiotensin II activates the Smad signalling pathway: Role in vascular fibrosis. J Hypertens 22:S351–S352
Rodriguez-Vita J, Sanchez-Lopez E, Esteban V, Ruperez M, Egido J, Ruiz-Ortega M (2005) Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation 111:2509–2517
Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH et al (2003) Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300:1394–1399
Rube CE, Uthe D, Schmid KW, Richter KD, Wessel J, Schuck A, Willich N, Rube C (2000) Dose-dependent induction of transforming growth factor beta (TGF-beta) in the lung tissue of fibrosis-prone mice after thoracic irradiation. Int J Radiat Oncol Biol Phys 47:1033–1042
Scotton CJ, Chambers RC (2007) Molecular targets in pulmonary fibrosis – the myofibroblast in focus. Chest 132:1311–1321
Shi Y, Massague J (2003) Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113:685–700
Sime PJ, O'Reilly KMA (2001) Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin Immunol 99:308–319
Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J (1997) Adenovector-mediated gene transfer of active transforming growth factor-beta 1 induces prolonged severe fibrosis in rat lung. J Clin Invest 100:768–776
Su Z, Zimpelmann J, Burns KD (2006) Angiotensin-(1–7) inhibits angiotensin II-stimulated phosphorylation of MAP kinases in proximal tubular cells. Kidney Int 69:2212–2218
Surjit M, Liu BP, Jameel S, Chow VTK, Lal SK (2004) The SARS coronavirus nucleocapsid protein induces actin reorganization and apoptosis in COS-1 cells in the absence of growth factors. Biochem J 383:13–18
ten Dijke P, Arthur HM (2007) Extracellular control of TGFbeta signalling in vascular development and disease. Nat Rev Mol Cell Biol 8:857–869
Tse GMK, To KF, Chan PKS, Lo AWI, Ng KC, Wu A, Lee N, Wong HC, Mak SM, Chan KF et al (2004) Pulmonary pathological features in coronavirus associated severe acute respiratory syndrome (SARS). J Clin Pathol 57:260–265
Uhal BD, Kim JK, Li XP, Molina-Molina M (2007) Angiotensin-TGF-beta 1 crosstalk in human idiopathic pulmonary fibrosis: autocrine mechanisms in myofibroblasts and macrophages. Curr Pharm Des 13:1247–1256
Wang JT, Chang SC (2004) Severe acute respiratory syndrome. Curr Opin Infect Dis 17:143–148
Waseda Y, Yasui M, Nishizawa Y, Inuzuka K, Takato H, Ichikawa Y, Tagami A, Fujimura M, Nakao S (2008) Angiotensin II type 2 receptor antagonist reduces bleomycin-induced pulmonary fibrosis in mice. Respir Res 9:43
Watanabe T, Barker TA, Berk BC (2005) Angiotensin II and the endothelium – diverse signals and effects. Hypertension 45:163–169
Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z (2005) Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-ss 1 – potential role in idiopathic pulmonary fibrosis. Am J Pathol 166:1321–1332
Wong CK, Lam CWK, Wu AKL, Ip WK, Lee NLS, Chan IHS, Lit LCW, Hui DSC, Chan MHM, Chung SSC, Sung JJY (2004) Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol 136:95–103
Wu Z, Yang LL, Cai L, Zhang M, Cheng X, Yang X, Xu J (2007) Detection of epithelial to mesenchymal transition in airways of a bleomycin induced pulmonary fibrosis model derived from an alpha-smooth muscle actin-Cre transgenic mouse. Respir Res 8:1
Wynn TA (2008) Cellular and molecular mechanisms of fibrosis. J Pathol 214:199–210
Xie LX, Liu YN, Fan BX, Xiao YY, Tian Q, Chen LG, Zhao H, Chen WJ (2005) Dynamic changes of serum SARS-Coronavirus IgG, pulmonary function and radiography in patients recovering from SARS after hospital discharge. Respir Res 6:5
Yoshida K, Kuwano K, Hagimoto N, Watanabe K, Matsuba T, Fujita M, Inoshima I, Hara N (2002) MAP kinase activation and apoptosis in lung tissues from patients with idiopathic pulmonary fibrosis. J Pathol 198:388–396
Zhao X, Nicholls JM, Chen YG (2008) Severe acute respiratory syndrome-associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor-beta signaling. J Biol Chem 283:3272–3280
Acknowledgments
The research in Y.G.C’s laboratory is supported by grants from the National Natural Science Foundation of China (30430360, 30671033), 973 Program (2004CB720002, 2006CB943401, 2006CB910102), and 863 Program (2006AA02Z172).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Zuo, W., Zhao, X., Chen, YG. (2010). SARS Coronavirus and Lung Fibrosis. In: Lal, S. (eds) Molecular Biology of the SARS-Coronavirus. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-03683-5_15
Download citation
DOI: https://doi.org/10.1007/978-3-642-03683-5_15
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-03682-8
Online ISBN: 978-3-642-03683-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)