
Abstract
Every living cell has to exchange molecules across the membrane for cellular functions. The hydrophobic or lipophilic molecules do not require energy for crossing the membrane. They can diffuse freely from higher to lower concentration till equilibrium is established. This process is called passive transport or diffusion.
You have full access to this open access chapter, Download chapter PDF
6.1 Introduction
Every living cell has to exchange molecules across the membrane for cellular functions. The hydrophobic or lipophilic molecules do not require energy for crossing the membrane. They can diffuse freely from higher to lower concentration till equilibrium is established. This process is called passive transport or diffusion. This is not true for polar or lipophobic molecules because of the nonpolar nature of membrane lipid bilayer. For polar molecules, the cell recruits mediator or transporter proteins in the membrane to facilitate their movement. If the direction of transport is from higher to lower concentration region, the transporter proteins do not use external energy and the process is called facilitated transport, because the energy needed for transport is dispensed by molecule’s own concentration gradient (unequal distribution across the membrane) in the favorable direction. The free energy is minimal in unequal distribution of polar molecules. If polar molecule is transported from lower to higher concentration region, against the concentration gradient, the energy has to be provided by external source to mediator protein to make the process spontaneous in the cell, and this process is called active transport. In the cell, the energy source is mostly adenosine triphosphate (ATP). The facilitated transport unlike diffusion has shown speed and specificity, saturation kinetics, susceptibility to competitive inhibition, and susceptibility to chemical inactivation. The active transport of polar molecules against concentration gradient requires energy for transport. A large group of transporters that hydrolyze ATP to transport molecules across the membrane are known as ATPases. These carrier ATPases are classified into five subclasses according to their structure and physiological functions as P-type ATPases, V-type ATPases, F-type ATPases, ecto-type ATPases, and A-type ATPases. P-type ATPase transporters are multidomain integral membrane proteins involved in the transport of cation and lipids. Phylogenetically according to their function, these P-type ATPases are divided into five major types and various subclasses. The vacuolar ATPases are ATP-dependent oligomeric protein proton pump, which regulate acidic pH in organelle compartment like phagosome and endosome for the separation of ligand from their receptors transport proton (H+) across the plasma membrane. The secondary transport, also known as ion-coupled transport involves the use of electrochemical potential generated by an ion transport for the co-transport of another ion against the gradient across the membrane. ATP-binding cassette (ABC) transporters, importers, and exporters, the newly identified integral proteins, comprise a large superfamily of integral membrane proteins with diverse functions. There are three major transporters which are known as flippases, floppases, and scramblases. The flippases are also known as P4-ATPases, which transport phospholipids to inner leaflet of membrane and have substrate specificity to phosphatidyl serine and phosphatidyl choline. Aquaporins (AQPs), the integral proteins of membrane, are small, hydrophobic, and homotetramer proteins, which are involved in bidirectional transport. The bacteria take nutrients from their surroundings by various modes of transport but the transport of sugars and its derivatives like sugar alcohols, amino sugars, glucuronic acids, and disaccharides is transported by group translocation with histidine heat resistance protein HPr as high-energy phosphate donor protein. The transporting sugar molecule is also phosphorylated during transport, which makes sugar negatively charged. The various organisms have specialized proteins of rhodopsin family to seize light energy for various physiological functions. The rhodopsin protein part, opsin, is covalently linked to retinal chromophore, which may be in trans- or cis-configuration. The animal rhodopsin (also known as Type II) are cell G-coupled receptors, while microbial rhodopsin (Type I) may act as ion pump, ion channels, sensors, photosensory receptors and regulator for gene expression and kinases. Pore-forming toxins (PFTs), the bacterial virulence factor, single major family of proteins, are secreted by gram-positive and gram-negative bacteria. PFTs may be present either in cytoplasm or in the membrane. The conformational change in cytoplasmic PFTs may translocate them to membrane in need of hour. Ionophores are natural and synthetic organic molecules of diverse type, which can transport cations by forming lipid ion-soluble complex. Each ionophore possesses a unique property of changing transmembrane ion gradient and electrical potential, synthesized by bacteria, and acts as protector against competing microbes. The bacterial and eukaryotic membranes are found to have special proteins in their outer membrane for transport, which are known as porins and form channels for various small size solutes, ions, and other nutrients. The protein ion channels are passive transporters, are made up of transmembrane, are selective for their substrate ions, and have much faster rate of transport than pumps. These channel proteins do not interact with transporting ions and have two conformations, closed and open, to regulate flow of ions. The trimeric ATP-activated channels are permeable to cations like sodium, potassium, and calcium. There are seven subclasses of the group, which are involved in various physiological functions like muscle contraction, neurotransmitter release and immune responses regulation. The GABA and glycine receptors are anion channels but inhibitory in nature. GABA receptors have two subdivisions as GABA receptors family of ligand-gated channel (ionotropic) GABAA and G receptor protein family (GABAB). The N-methyl-d-aspartate (NMDA) receptors are known as glutamate-gated cation channel for calcium permeation. The tetrameric ionotropic receptors include NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate receptor as per their synthetic ligand binding. The voltage-gated ion channels open or close in response to change in voltage and the unit of membrane potential. In excitable cells like nerve and muscle, the chemical or electrical signaling is initiated through voltage-dependent sodium or calcium influx. The K2P channels or voltage insensitive channels formerly referred to as leaky or resting channels, are ubiquitously found in all organisms. The functionally active K2P channel has two subunits, and each subunit is made up of two pore regions (P1 and P2), four transmembrane domains (M1–M4), and two characteristic extracellular helices Cap C1 and C2. This chapter explains all the mechanisms of membrane transport.
6.2 Passive Diffusion
While osmosis is the movement of solution from high concentration to low concentration without transporters across the lipid bilayer, the diffusion is a type of passive transport where small lipophilic or hydrophobic solute moves from higher to lower concentration. The membrane is made up of lipid bilayer to have hydrophobic interior in membrane, which is selectively permeable to small hydrophobic molecules but not to large polar ones. The passive diffusion of charged species across the membrane depends upon the concentration and also on charge of particle Z and the electrical potential difference across the membrane \(\Delta \psi\). The diffusion of particle across the membrane is determined by Fick’s law of diffusion and affected by magnitude of concentration gradient, molecular weight of solute, distance, permeability, and surface area of membrane. The diffusion coefficient of any solute molecule of Fick’s law is proportionality factor, which can be defined as mass of solute diffusing through a unit area in a unit time (unit is square meter per second). The diffusion coefficient depends on the size, temperature, charge on the molecule because of difference between proton and electron and polarity (uneven distribution of charge in the molecule) (Fig. 6.1).

Permeability coefficients of different molecules in the membrane. The nonpolar molecules can diffuse easily across the membrane. http://book.bionumbers.org/wpcontent/uploads/2014/08/650-f1-PermeabilityAxis-13.png
According to Fick’s law, for membrane area A, thickness X, and concentration gradient C2 (higher) and C1 (lower) across the membrane, the diffusion rate will be proportional to probability coefficient and can be written as follows:
The rate of diffusion is also proportional to partition coefficient K.
So,
where D is diffusion coefficient.
Equation (6.3) explains that the rate of diffusion is directly proportional to both partition coefficient and diffusion coefficient and inversely proportional to thickness of the membrane X; the thickness is constant factor for membrane; that’s why diffusion largely depends on partition coefficient. The more hydrophobic molecule will move much faster (Fig. 6.2).

Diffusion of polar and nonpolar molecules in the artificial phospholipid bilayer. The small nonpolar molecules move much faster than polar and large molecules
As depicted in Fig. 6.1, the hydrophobic nonpolar molecule diffuses much faster than polar molecule. Among the polar molecules, anionic molecules have higher permeability than cationic molecules. The gases like O2 and CO2 are nonpolar and hydrophobic, so they easily diffuse. Glycerol also diffuses across the membrane, but if this is phosphorylated, then this becomes polar. The urea (NH2CONH2) is less hydrophobic than diethyl urea (C2H5NHCONHC2H5), so urea diffuses slower than diethyl urea.
The diffusion of molecules and gases from mother to fetus has been observed, and another molecule sodium thiopental is observed to diffuse through blood–brain barrier.
6.3 Facilitated Transport of Glucose
Facilitated transport is mediated by transporter protein, which is found in the membrane without using external source of energy. The transport occurs from higher concentration to lower concentration. In mammal’s erythrocytes, the glucose is transported by facilitated diffusion by glucose transporter protein. The facilitated transport unlike diffusion has shown speed and specificity, saturation kinetics, susceptibility to competitive inhibition, and susceptibility to chemical inactivation (Fig. 6.3).

Cellular uptake of glucose mediated by GLUT protein exhibits simple enzyme kinetics and greatly exceeds the calculated rate of glucose entry by simple diffusion
The permeability coefficient of the facilitated transport of glucose is much higher than permeability coefficient (2.6 × 10−8 cm/s) of diffusion. The glucose transporter proteins are divided into two major families like Na+/glucose cotransporter (SGLTs) and facilitative glucose transporter (GLUTs). The SGLT is found in specialized epithelial cells of intestine, proximal tubule of kidney, trachea, heart, brain, testis, and prostate. The Na+/glucose cotransporters (SGLTs) are involved in secondary transport of glucose in intestine and nephrons. The 12 members of SGLT class are identified; out of them SGLT1, SGLT2, SGLT4, and SGLT5 function as sugar transporter and SGLT3 acts as glucose sensor. SGLT2 is present in the kidney, brain, liver, thyroid, muscle, and heart. The facilitative glucose transporter (GLUTs) has 14 members, and 11 out of 14 transport sugars. GLUT1 is present in erythrocytes, fetus placenta, brain, and kidney but low in liver and muscles. GLUT2 expressed in liver maintains glucose homeostasis. GLUT3 is neuron-specific glucose transporter with Km of around 2 mM for glucose. GLUT4, a high-affinity, insulin-responsive transporter with approximately 5 mM K m, is expressed in adipose tissue and muscle tissue. In the process of insulin-stimulated GLUT4 translocation, the various proteins of signal pathway also participate. The high-affinity enzyme GLUT1 has Km for glucose around 3–7 mM lower than blood glucose level (5–7 mM), hence transports glucose at significant level even in hypoglycemic conditions. The human erythrocyte glucose transporter (GLUT1) is a type III transporter, having 492-residue glycoprotein with 12 transmembrane (M1–M12) spanning α helices (Mr-45,000). The N-terminal and C-terminal are located in cytoplasm. 33 amino acids in the extra cellular loop between M1 and M2, have n-linked glycosylation sites. The large 65 hydrophobic amino acids are present between M6 and M7. The 3, 5, 7, 8, and 11 amphipathic helices are involved to form hydrophilic channel for glucose transport (Fig. 6.4).

Structure of GLUT1 with 12 transmembranes, and the alpha-helices 3, 5, 7, 8, and 11 are involved to form hydrophilic channel for glucose
The glucose transporter accounts for 2% of erythrocyte membrane proteins and runs as band 4.5 in SDS-PAGE gels of erythrocyte membranes. It is generally not visible on the gel because the heterogeneity of its oligosaccharides makes the protein band to diffuse (Fig. 6.5).

Identification of RBC proteins by SDS
The glucose concentration is maintained at 4 mM for all metabolic purposes inside the cell. The process of glucose transport can be compared to an enzymatic reaction, where the glucose concentration outside the cell is the substrate (S out), glucose concentration inside the cell is the product (S in), transporter T is the enzyme, initial velocity is (V 0) and (Kt) is the transport constant like Michaelis constant. The graph between velocity of transport and extracellular glucose concentration [S]out shows saturation kinetics. V max and Kt can be calculated from this double reciprocal plot between 1/V 0 and 1/S out when [S] = Kt, and the rate of uptake is 1/2 of V max. The rate equations for this process can be derived like enzyme-catalyzed reactions from Michaelis–Menten equation:
The glucose molecule does not undergo any chemical change during transport across the membrane making this a reversible process and helps to achieve equilibrium much faster in the presence of transporter GLUT1 transporter. The transporter protein undergoes a change in conformation on binding to glucose. It has a Kt of 1.5 mM for d-glucose (Fig. 6.6).

A saturation curve of glucose between velocity and substrate concentration B, the line weaver burk double reciprocal plot of glucose between 1/v and 1/S
6.4 Facilitated Chloride–Bicarbonate Transport
The carbon dioxide formed by cellular respiration of tissues is transported to lung as bicarbonate through erythrocytes and plasma. The hydration of CO2 by carbonic anhydrase in erythrocytes forms bicarbonate, which is exchanged with chloride. The anion transporter involved in facilitated transport across the erythrocyte membrane is high-capacity transporter as the diffusion rate of chloride/bicarbonate is never rate limiting for CO2 transport to lungs. This anion transporter constitutes 30% of erythrocyte membrane. The two identified families of anion exchanger proteins are known as SLC/4A and SLC 26A. The chloride–bicarbonate anion transporter belongs to SLC 26A family. SLC/4A has Na+/bicarbonate cotransporter. The anion exchanger can transport bicarbonate in both directions. These transporters regulate pH of the cell, volume, and acid–base secretion.
The carbonic anhydrase enzyme (pKa of 6.2, close to cellular pH) along with CO2/HCO3 equilibrium acts as a buffer and maintains pH in the cell (Fig. 6.7).

Anion exchanger protein in RBC. Box highlights transmembrane domains. Residue E681 participates in permeability barrier
The human chloride–bicarbonate anion exchanger protein has 911 residues with N-terminal domain, transmembrane, and C-terminal domain. The N-terminal and C-terminal are cytosolic. This comprises 50% of RBC protein. There are 12–14 transmembrane domains involved in transport. The protein is glycosylated at 642 asparagine residue of extracellular domain four. The C-terminal domains of 40 amino acids interact with carbonic anhydrase enzyme (transport metabolon).
In erythrocyte, carbonic anhydrase reaction forms bicarbonate.
During mitochondrial respiration, CO2 is diffused in erythrocytes via capillaries, where it is converted into bicarbonate reaction as shown here. The bicarbonate formed in tissue erythrocytes is released in plasma for lungs. The process is reversed in lungs; the bicarbonate is transported back to lung erythrocytes, which is converted there into CO2 by carbonic anhydrase II isoform of lung and CO2 is exhaled from lung by diffusion. In the exchange of one bicarbonate ion, chloride ion also moves in opposite direction; that’s why this anion transporter is called electroneutral transport (Fig. 6.8).

Facilitated chloride bicarbonate anions transport in erythrocytes of tissues and lungs
This anion transporter increases blood exchange capacity for CO2 and provides flexibility of erythrocytes by interacting with RBC cytoskeleton. This transporter may be dimer or tetramer in erythrocytes. The N-terminal interacts with ankyrin, β-spectrin, 4.1 and 4.2 band proteins of RBC. The mutation in RBC anion exchanger results in defective interaction with cytoskeletal proteins to make RBC osmotic fragile and causes disease such as hereditary spherocytosis. In kidney, the anion exchanger protein controls acid–base homeostasis, electrolyte, and fluid concentration.
6.5 Primary Active Transport
The carrier protein as discussed earlier can be involved in active or passive transport. The active transport of polar molecules against concentration gradient requires energy for transport. A large group of transporters that hydrolyze ATP to transport molecules across the membrane are known as ATPases. These carrier ATPases are classified into five subclasses according to their structure and physiological functions as P-type ATPases, V-type ATPases, F-type ATPases, ecto-type ATPaeses, and A-type ATPases.
-
1.
P-type ATPases (E 1 –E 2 ATPases): These ATPases transport various cations and lipids across the cellular and organelle membrane to regulate cell functions. The various cations may be H+, Na+, K+, Ca2+, Cu2+, Cd2+, Hg2+, Zn2+, Ni, and Pb2+. The phylogenetic study has divided this group into five major types and more than 11 subtypes.
-
2.
E-type ATPases: These are cell surface transporters involved in extracellular transport, which can use nucleoside diphosphates and nucleosides triphosphates including extracellular ATP. These transporters depend upon Ca2+ and Mg2+ and are insensitive to specific inhibitor of P-type. Examples are ecto-ATPase and animal ecto-apyrase. The E-type ATPases participate in termination of purinergic receptor-mediated responses. They are also involved in cell adhesion processes and are important for the maintenance of hemostasis in the cardio vasculature.
-
3.
F-type ATPases (F 1 –F 0 ATPases): These ATPases are involved in ATP synthesis and localized on bacterial membrane, thylakoid membranes of chloroplasts, and inner membranes of mitochondria.
The detail of these ATPases is discussed in Chap. 8.
-
4.
V-type ATPases (V 1 –V 0 ATPases): These ATPases act as proton pump and are found on various organelle membranes to regulate acidic pH in organelles.
-
5.
A-type ATPases (A 1 –A 0 ATPases): These ATPases are present in archaeal bacteria and in some other prokaryotes. They functionally resemble to F-type ATPases and structurally to V-type ATPases.
6.6 P-Type (E1–E2) ATPases
P-type ATPase transporters are multidomain integral membrane proteins involved in transport of cation and lipids. Phylogenetically according to their function, these P-type ATPases are divided into five major types and various subclasses (Fig. 6.9).

Phylogenetic data of the P-type ATPase. Subfamily division according to their function and ion binding specificity type IA, bacterial Kdp-like K+-ATPases; type IB, soft-transition-metal translocating ATPases; type IIA, sarcoplasmic reticulum (SR) Ca2+-ATPases; type IIB, plasma membrane Ca2+-ATPases; type IIC, Na+/K+-ATPases and H+/K+-ATPases; type IID, eukaryotic Na+-ATPases; type IIIA, H+-ATPases; type IIIB, bacterial Mg2+-ATPases; type IV, “lipid flippases”; and type V, eukaryotic P-type ATPases of unknown substrate specificity. Colour coded by species: green, genes from Arabidopsis thaliana; orange, C. elegans; grey, E. coli; dark blue, Homo sapiens; light blue, Methanobacterium thermoautotrophicum; yellow, Methanococcus jannaschii; purple, Synechocystis PCC6803; and red, Saccharomyces cerevisiae
The P-type ATPase or pump is a 70–150 Kd protein having approximately around 900 amino acids. P-type ATPases may have maximum 10 hydrophobic transmembranes from M1–M10 with their N-terminal and C-terminal of proteins in cytoplasm. The cytoplasmic domain is highly conserved and inserted between M2 and M3 and M4–M5. These proteins have characteristic conserved amino acids DKTGTLT, where aspartic acid (D) is reversibly phosphorylated during catalytic cycle. The five types of P-type ATPases are divided as Type I, Type II, Type III, Type IV, and Type V.
Type I P-type ATPases are bacterial ion pump like Escherichia coli–Kdp ATPases, which are made up of four different subunits with six transmembrane domains namely, Kdp F, A, B, and C. The Kdp B is the catalytic and ion-transporting subunit of the pump. Type IB ATPases of single peptide with eight transmembranes like ZntA and CopA physiologically detoxify bacterial cell by throwing out toxic metals.
Type II P-type ATPases are majorly involved in membrane potential regulation in the cell. They are present ubiquitously in all the cells like Na+/K+ ATPases and H+/K+ ATPases. Type IIA is most studied and diversified class that includes sarcoplasmic reticulum Ca2+ ATPase pump (SERCA). This calcium ATPase transports two calcium ions from muscle to lumen of endoplasmic reticulum at the expanse of one ATP and in exchange of two or three hydrogen ions. The muscle contraction is controlled by Ca2+ ATPases because the sudden calcium increase in muscle cell results in muscle contraction. The Type II A calcium ATPases is inhibited by phospholamban, which is active in dephosphorylated form and blocks calcium entry by interfering in M2 transmembrane movement. The calcium ATPases are inhibited by thapsigargin. Type IIB includes plasma membrane calcium ATPases or pump (PMCA). The Type IIB ATPases are regulated by their C-terminal or N-terminal calmodulin-binding domains. The Type IIC ATPases are heteroligomers with α- and β-subunits. The sodium–potassium ATPases (Na+/K+ATPase) belong to this class. The α-subunit with maximum ten transmembranes (M1–M10) is catalytic unit with autophosphorylation site and ion binding site, while β-subunit, glycosylated single transmembrane of 55 Kd, is required for insertion and assembly. The renal Na+/K+ ATPase has additional γ-subunit with FXYD motif (Fig. 6.10).

Structure of sodium–potassium ATPases. These ATPases are composed of two subunits (α and β). The α-subunit (113 Kd) binds ATP and ions (Na+ and K+) and contains an autophosphorylation site (P) but some isoforms like renal sodium–potassium have extra γ subunit with FXYD2
The FXYD proteins act as regulator and stabilizer. The FXYD motifs provide stability to pump. The sodium–potassium pump is inhibited by digitalis glycosides and ouabain, and H+/K+ ATPases are inhibited by omeprazole, an antiulcer drug, so these pumps are the site of drug action also. The Na+/K+ ATPases transport three Na+ ions out of the cell and two K+ ions inside the cell per catalytic cycle. The Type IID ATPases are eukaryotic Na+/K+ ATPases.
The Type III ATPases are prominently found in fungi and plants, where these pumps maintain intracellular pH of 6.6 against extracellular pH of 3.5 and maintain membrane potential in the similar way of animal Na+/K+ pump. The H+ ATPases are regulated by C-terminal autoinhibitory amino acids. Fusicoccin is a fungal toxin, which activates H+ ATPases irreversibly.
Type IV and Type V ATPases’ role is not very clear but structurally they resemble to Type I and are found in eukaryotes. They are also known as flippases, which transport phospholipids from outer leaflet to inner leaflet and maintain lipid asymmetry of the membrane. The erythrocyte membrane has Mg2+-dependent flippases. These ion pumps may work in correlation with other lipid transport proteins to maintain asymmetry of membrane.
6.6.1 Structure of P-Type ATPases
X-ray crystallography, NMR spectroscopy and electron cryo-microscopy (EM) studies on various types of P-type ATPases especially, calcium ATPases and sodium–potassium ATPases revealed that P-type ATpases have four domains with conserved sequences, namely, actuator A, membrane M, nucleotide-binding domain NBD, and phosphorylation P-domain. The variation of amino acids has been observed only in extracytoplasmic loop.
6.6.2 Phosphorylation Domain
The most conserved phosphorylation domain is the catalytic domain with DKTGTLT signature motif, where aspartic acid (D) is phosphorylated and dephosphorylated during catalytic cycle. It has Rossmann fold (characteristics of many nucleotides proteins) made up of central seven-stranded β-sheet flanked by α-helices, including the cytoplasmic end of M5.
6.6.3 Nucleotide-Binding Domain
The nucleotide-binding domain inserts into P-domain through conserved hinge region of two antiparallel peptides. Seven strands of β-sheet in central have lysine 515 for nucleotide binding. In Na+/K+ ATPases—N-domain phenylalanine clustering is observed whereas acidic residues are rich in calcium ATPases. This might be site for regulator interaction, as observed with EM. In the nucleotide binding site, adenosine base and phenylalanine 475 interaction forms hydrophobic stacking and three phosphate group bulge out in solution so that γ-phosphate could reach to P-domain aspartic acid for phosphorylation.
6.6.4 The Actuator Domain
The N-terminal actuator domain is highly conserved. Similarly like P-domain, A-domain has two unequal divisions by M1 and M2 helices hairpin structure. The larger part has β-sheet secondary structure with conserved TGE sequence. The TGE sequence interacts with phosphorylation site during transport cycle. Some of ATPases like fungal H+-ATPases have extra-long amino-terminal amino acids fused with actuator domain, which might play role in substrate sensing or in regulation.
6.6.5 Membrane Domain
The membrane domain is the largest domain of approximately 405 amino acids with ten transmembrane segments to enclose ion binding site. This domain is less conserved and is directly connected to catalytic core through M4–M5 and M6–M8 polar ionic side chains. As observed in calcium ATPases, the M6 aspartic acid 800 and M8 glutamate 908 acidic side coordinate Ca2+; the other amino acids such as valine 304, alanine 305, and isoleucine 307 of M4 also participate in ion binding. M is the flexible region, which is arranged and rearranged during transport cycle for accommodating different sizes and charge ions and for catalysis (Fig. 6.11).

Alignment of various domains in sarcoplasmic reticulum Ca2+-ATPase. The conservative substitutions are shown here in red. The most conservative residues are found in the phosphorylation (P)- and actuator (A)-domains, fewer in the nucleotide-binding (N)-domain, and hardly any in the 10 membrane-spanning helices
6.6.6 Transport Cycle of P-Type ATPases
The catalytic cycle of ATP binding, phosphorylation, changes in conformation by arrangements of M-domain helices, hydrolysis of ATP for energy are same in all P-type transporters. That’s why P-, N-, and A-domains have conserved residues but M-domain with variable residues regions with ion binding site may change to accommodate various ions. The cycle starts with loading of ion 1 (X+) to high-affinity binding site and releasing of extracellular ion 2 (Y+) in cytoplasm through cytoplasmic access channel. The interaction of ion 1 (X+) with polar group of amino acids near binding site at P-domain twists M4, M1, and M6 helices to move in E1 state. Mg2+/ATP binding to NBD domain brings this close to P-domain through slight movement of β-strands. These movements result in correct positioning of γ phosphate group of ATP to P site, and E state of ATPase is changed to E1~P state. In E1~P state, ion1 (X+) is trapped in protein, which can be visible from any side of membrane. When ion1 (X+) binding is complete, the phosphorylation takes place on aspartic acid residue of P-domain because the energy released by ion binding to E1 ATPase favors the inclination of P-domain to bring more closure γ phosphate group to aspartic acid for reaction (Fig. 6.12).

Transport cycle of P-type ATPases. In the E1 conformation, ion 1 (X+) binds to its high-affinity site in the membrane (M)-domain through cytoplasmic side. The binding of X+ ion initiate phosphorylation of aspartic residue in E1 state (P-domain by ATP). Phosphorylated E1P state transporter still is in same state. During transition of E1 to E2 change in conformation (regulated step), the P-domain rearranged itself for bringing TGE loop near to phosphorylation site, which stabilizes phosphoryl group binding and ADP dissociates. The A-domain M1 and M2 segments movement closes access to cytoplasmic ion access channel (light gray). The P-domain rotation disturbs the high-affinity X+ binding site through its attachment to helices M4–M6 (dark gray). X+ is delivered to the outside (extracellular/lumenal side) through an exit channel. The ion binding site of E2 P now has a high affinity for second ion 2 (Y+), which may bind from the outside. The aspartic acid is dephosphorylated leaving enzyme into E2 conformation. Mg2+ and inorganic phosphate (Pi) are dissociated, and the enzyme reverts back to the E1 state, in which Y+ is released into the cell. The ATPases are ready for new cycle
The reaction may be explained as nucleophilic attack on aspartic acid in P-domain by γ \({{phosphate}}\) group. This may be the reason that sodium ATPase needs three Na+ ions in comparison with two Ca2+ ions in Ca2+ ATPases for P-domain movement. A-domain rotates parallel to membrane, and TGE loop becomes closure to phosphorylation site. The E1~P state changes to E2~P state. Simultaneously, M1, M2, and M3 helices of A-domain because of their rearrangement induced by phosphorylation may close polar ion access channel from cytoplasmic site. This might be understood that the inclination of P-domain induces the movement of helices in such a way that E2~P state has low affinity to ion1 (X+) and leaves this to extracellular site or into lumen of ER. The E2~P state has high affinity for ion 2 (Y+), and binding to this ion may induce the hydrolysis of phosphorylated aspartic acid. The TGE loop interaction with Mg2+ ion will leave phosphate group unprotected, and this may result in hydrolysis by water. After this hydrolysis, E2 transforms into E2 state with open binding site in cytoplasm for next cycle of transport.
6.7 Vacuolar [V-Type] ATPases
The vacuolar ATPases are ATP-dependent oligomeric protein proton pump, which regulate acidic pH in organelle compartment like phagosome and endosome for the separation of ligand from their receptors and transport proton (H+) across the plasma membrane. The V type ATpases activity can be regulated by reversible disulphide bond formation between conserved cysteine residues at catalytic site. Glutamate uptake in presynaptic vesicle is found to be dependent on V-type ATPases. The V-type ATPases regulate plasma pH by exporting proton through renal intercalated cells of kidney. The mutation in V-type ATPases may cause renal tubular acidosis. The vacuolar ATPases also regulates acidic pH in osteoclasts for bone degradation, and the genetic defect in V-type ATPases may cause osteoporosis. V-type ATPases are present at the surface of neutrophil and macrophage for maintaining pH. The sperm maturation and storage also need V-type ATPases in epididymis of testis. The cancer cells are also found to manipulate V-type ATPases. The V-type ATPases are potential target for drug in various diseases. The prerenin receptors’ study has indicated the role of renin/angiotensin signaling in V-type ATPases’ regulation. The V-type ATPases are oligomeric protein complex with two different domains, which are known as peripheral (V 1 ) of eight subunits and integral (V 0 ) of six subunits. While peripheral proteins (V1) carry out ATP hydrolysis, the V0-domain of integral proteins transports proton ions. The peripheral proteins V1 are designated in bold alphabet like V 1 A, B, C, D, E, F, G, and H, and integral membrane proteins are known as a, b, c, c”, d, and V 0 e. The various isoforms of these subunits are found in various tissues and organs. The peripheral proteins are present in stoichiometry concentration of A3 B 3 C 1 D 1 E 2 F 1 G 2 H 1–2 . The catalytic domain of vacuolar ATPases contains heterohexamer of alternate A- and B-subunits with the ATP binding site at the interface of A- and B-subunits. Three subunits of A contribute majorly to three catalytic sites, and B-subunits are regulatory in functions. The remaining peripheral proteins form central and peripheral stalk, which is linked to V1- and V0-domains. C-, E-, G-, and H-subunits form the peripheral stalk. The V0-subunits are present in the ratio of a 1 d 1 e n c 4–5 \({\mathbf{c}}_{{\mathbf{1}}}^{\prime}\) and \({\mathbf{c}}_{{\mathbf{1}}}^{\prime \prime}\) (n is variable number of e). The subunits c, \({\text{c}}^{\prime}\), and \({\text{c}}^{\prime \prime}\) have hydrophobic residues in high concentration and have one buried carboxyl group in one of the transmembranes as TM4 (for c and \({\text{c}}^{\prime}\)) and TM3 (for \({\text{c}}^{\prime \prime}\)) for proton translocation. These buried carboxyl groups may be reversibly protonated during proton translocation. These hydrophobic subunits with single copy of \({\text{c}}^{\prime}\) and \({\text{c}}^{\prime \prime}\) and four copies of c form proteolipid ring. The hydrophilic subunit d at the cytoplasmic side interacts with V1 subunits (D and F) of the central stalk complex, and this whole complex is referred as rotatory complex (Fig. 6.13).

Structure of V-type ATPases with various isoforms in human. A-subunit catalyzes ATP hydrolysis. 3 A and 3 B-subunits of V 1 form heterohexamer. The conformational changes in the AB heterohexagon promote turning of the central rotor and the ring of c- and c″ (b)-subunits. https://www.hindawi.com/journals/njos/2014/675430/fig2
The subunit a of V 0 with eight transmembrane domains has buried arginine amino acid in seventh transmembrane domain, which participates in proton translocation by forming two hemichannels (one toward cytoplasmic side and other in opposite side). The cytoplasmic proton enters through cytoplasmic hemichannel of subunit A, which may protonate buried carboxyl group of V0-subunit. The ATP hydrolysis by A-subunit induces conformational change in heterohexamer of AB resulting in rotation of central motor with D-subunit as axle of the motor. F is regulatory unit. V 0 d-subunit couples this rotation to ion-transporting proteolipid ring of b- and c- subunits. The b-, c-, and a-subunits form channel for proton transport. The proton moves with rotation of b- and c-subunits. The V-type ATPases function through rotatory mechanism, where V1-induced ATP hydrolysis results in rotation of central rotatory domain with ring of hydrophobic subunits (c, \({\text{c}}^{\prime}\), and \({\text{c}}^{\prime \prime}\)) in relation to other subunits, which causes ion transport.
This proton remains to interact with carboxyl group because of only polar group in the hydrophobic surroundings of V0-subunits. When ATP hydrolysis induces conformation change by rotation, the protonated carboxyl group is exposed to luminal hemichannel. These carboxyl groups interact with buried arginine residue of a-subunit and stabilize carboxyl group as carbonate ion, so proton gets disassociated and released into the luminal hemichannel. The continued movement of charged carboxyl group because of changes in conformation by rotation of subunit a places it once again in contact with the cytoplasmic hemichannel, where it is again available for protonation.
Regulation of V-type ATPases can be observed through the following different ways either induced or natural. The V-type ATPases may be regulated by (1) reversible dissociation of V1 and V0-subunit, (2) by changing or uncoupling of proton transport and ATP hydrolysis, (3) by reversible disulfide bond formation between cysteine 254 and cysteine 532 of catalytic subunit A, (4) by modulation proton pump density as in apical membrane of renal intercalated cell of kidney proton pump activity is regulated by reversible fusion of the pump with apical cells induced by bicarbonate-sensitive adenylate cyclase. The net pH of a compartment is the result of active proton transport and passive proton leakage. If passive proton ion diffusion is controlled, proton pump automatically will be regulated (Fig. 6.14).

Source Cipriano et al. (2008)
V-type ATPases may be regulated by a reversible disulfide bond formation between conserved cysteine residues of catalytic A-subunit, b controlling proton ion pump density through fusion with membrane, c regulation of other transporters for Cl− or H+, d reversible dissociation of V1 and V0 domains and e uncoupling ATP hydrolysis and transporting pump subunits.
F-Type ATPases are discussed in detail in Chap. 8.
6.8 Secondary Transport
The secondary transport is also known as ion-coupled transport because the electrochemical potential generated free energy of an ion transport which is used by transporters for other substrate transports across the membrane. These transporters ubiquitously found in all cells constitute a major facilitator superfamily (MFS) or solute carrier group of transporters (SLC). More than 4000 of these transporters are involved in lipid, amino acids, sugar transport, and antibiotics. The important examples may include Na + /glucose (SGLT1) transporter present in kidney proximal tubule and epithelial cells of intestine. Oligonucleotide/proton transporter (facilitated proton transport and uphill peptide transport), proton/neurotransmitter transports in synaptic vesicle membrane of neuron (neurotransmitter transport against concentration gradient by using electrochemical energy generated by proton transfer), lactose permease LacY in bacteria, sodium taurocholate cotransporting peptide (OATP), apical sodium-dependent bile acids (APSBT). The major facilitator superfamily transporters have 12–14 transmembrane segments and transport substrate by alternating conformations. The bacterial lactose secondary transporters (LacY) and sodium glucose transporters (SGLT1) in human intestines are two well-explored transporters to understand secondary transport mechanism, which is discussed here.
6.8.1 Secondary Transport of Disaccharide Lactose by Lactose Permease (LacY)
Lactose permease (LacY) transporter of E. coli bacteria has selective affinity for disaccharides containing a d-galactopyranosyl ring, as well as d-galactose and symport d-galactopyranoside and hydrogen ion together in cell (symport). LacY protein is an integral protein (374 amino acids) of 12 transmembrane helices with a connecting loop between two pseudo-symmetrical six α-helice (7–194 and 214–401 amino acids) bundles. These α-helices form a central hydrophomation. The other conformation with open cavity toward periplasmic side is called outward-facing conformation. This alternate conformation of transporter (involved helices II, IV, V in N-terminal and VII, X, XI in C-terminal) exposes bound sugar and proton to cytosol and periplasmic side alternatively; the basic principle of this type secondary transport is the conformation change in transporter by binding with substrate. The sugar-interacting residues are present in N-terminal region and the proton-interacting amino acids are present in C-terminal side (Fig. 6.15).

a Structure from top of lactose permease. b Cytosolic view of lactose permease with inward open cavity
The mutagenesis study has identified the role E126 of helix IV, R144 and W155 of helix V in substrate recognition and binding and R302 of helix IX, E325 and H322 of helix X in proton translocation. E269 is required for sugar identification and proton translocation.
LacY has shown affinity toward numbers of d-galactopyranosyl disaccharide and with d-galactose also but not to d-glucose. The 4-hydroxyl group is found to have role in discrimination.
The X-ray crystallography of LacY transporter has explained the mechanism. The transport cycle starts with inward open conformation. In the absence of sugar, lactose permease transporter is closed from periplasmic side. The binding of d-galactosyl pyranoside to LacY transporter induces changes in protein to open periplasmic side by closing cytosolic inward open cavity. The deprotonation of E325 causes closing of inward cavity and opening of periplasmic cavity. The membrane potential has no effect on substrate binding. The five residues R144, W151, E269, N272, and H322 bind directly to substrate. The E126 and Y236 stabilize R144 and H322. All these together form substrate binding site in the center of protein. The translocation of substrate may be induced by hydrophobic interaction of W151 indole ring and d-galactosyl pyranoside ring. The binding of this nonpolar substrate to transporter induces major rearrangement of helices in the vicinity of substrate binding site for perfect accommodation. The substrate binding may lower down activation energy for transition between inward open and outward open conformation of LacY. In LacY, the galactose binding induces major conformational change in N-terminal six helices bundle (Fig. 6.16).

Lactose permease mediated lactose/H symport. In the absence of substrate, LacY does not translocate proton in the presence of electrochemical gradient of membrane. a Free energy released from the downhill movement of H is coupled to the uphill accumulation of lactose. Substrate binding generates electrochemical proton gradients. The polarity depends upon substrate concentration gradient direction. b, c Substrate gradients generate electrochemical H gradients, the polarity of which depends upon the direction of the substrate concentration gradient
The substrate binding and dissociation from transporter promote transition between inward- and outward-facing conformations. The lactose/proton symport has same mechanism in active or facilitative transport for galactose and proton but with the difference in rate limiting steps. The deprotonation step is rate limiting in favorable concentration gradient downhill transports, but in uphill transport, conformation changed induced protonation is rate limiting.
The lactose permease (LacY) transfer potential energy, preserved in proton gradient facilitates galactose transport down the concentration gradient. Hence, the transport is typically a thermodynamically driven and kinetically regulated secondary transport.
6.8.2 Sodium/Glucose Secondary Transport
There are various ways of glucose transporters as discussed earlier. The sodium/glucose (SGLT1) transporter is present in epithelial cells of human intestine and also found on incretin (intestinal hormone, which enhances insulin secretion)—secreting vesicles and also in S2 and S3 segments of in kidney proximal renal tubule. SGLT1 has 14 transmembranes (Fig. 6.17).

Source Wright et al. (2011)
Human glucose/sodium transporter (hSGLT1) structure with 14 transmembrane domains.
SGLT1 has specificity for glucose and galactose. In intestine, the sodium/glucose transporters translocate one molecule of glucose per two sodium ions across the epithelial membrane of intestine by using electrochemical gradient of another ion (sodium/potassium). The electrochemical potential is generated by another sodium/potassium pump located in basolateral membrane of these polarized cells of intestine. The another sodium/potassium ATPase pump is present in basolateral membrane to generate electrochemical gradient along with an independent glucose transporter, which also participates in translocation of glucose molecule from intestine to blood vessels. The detail mechanism of secondary transport is discussed above. The sodium–potassium ATPase pump at basolateral membrane transports two potassiums inside the cell and three sodium ions out in blood in ATP-dependent manner. The electrochemical gradient energy generated by this transport is used by SGLT1 transporter in epithelial cell of intestine to import glucose against concentration gradient. When glucose concentration is accumulated in cell, the other facilitator transporter of glucose transports accumulated glucose from cell to blood (Fig. 6.18).

Summary of glucose transport in human intestine and then to blood vessels by secondary transporter sodium/glucose (SGLT1). The mechanism is discussed in text
6.9 ABC Transporter
ATP-binding cassette (ABC) transporters, importers, and exporters, the newly identified integral proteins, comprise a large superfamily of integral membrane proteins with diverse functions. A total of 48 transporters are identified in human. In bacteria 1% and in archaeabacteria 3% of the genome codes for ABC transporters. They might transport various substrates like amino acids and peptides, monosaccharide, polysaccharides, nucleosides, oligonucleotides, vitamins, coenzymes, and different types of lipid. The ABC transporter proteins are classified on the basis of highly conserved approximately 180 amino acid residues with three motifs like Walker A/P-loop (12 amino acids), a signature motif/C-loop (LSGGQ), and the Walker B motif (five amino acids), which participate in ATP hydrolysis.
ABC exporters are physiologically involved in multidrug resistance and detoxification, while importers play role in nutrient uptake and also are used by some prokaryotic pathogens to show pathogenicity and virulence in plants, prokaryotes, and archaea. The ABC exporters are also known as multidrug resistance transporters because of their role in detoxification. The other distinct classes of energy-coupling factor (ECF) transporters are also as class III ABC importers. The ABC exporters and importers are further classified into subclasses as Type I, II, and III. Functionally, these all transporters may be divided into two major domains known as nucleotide-binding domain (NBD) and transmembrane domain (TMD). The NBD and TMD can be further divided into NBD1 and NBD2 and TMD into TMD1 and TMD2. These domains may be separate, dimer, or fused peptides. In eukaryotes, the ABC transporters are mostly multifunctional with single peptide with four domains but the assemble to form dimers or heteromers. In bacterial ABC transporters NBD and TMD may be separate, dimer or fused. In some bacteria an extra subunit of 30–35 Kd is found for binding to substrate and delivery to TMD. The bacterial ABC transporters may be localized in periplasm like gram-negative bacteria or may be membrane bound like gram-positive bacteria (Fig. 6.19).

a The three classes of ABC transporters, based on numbers of transmembrane A full transporter such as multidrug resistance MDR1 and multidrug-resistance-associated protein 4 MRP4 have 12 transmembrane domains and two ATP binding sites. b The structures of MRP1, 2, 3, and 6 of 17 transmembrane, with two ATP-binding regions, an additional domain of five transmembrane segments at the amino-terminal end. c The “half-transporter” with six transmembrane domains and one ATP-binding region. The ATP-binding camay be on the carboxy-terminal (C) or N-terminal
As mentioned above, the characteristic ABC transporters conserved signature motif like Walker A (GKT/S for phosphate binding domain), LSGGQ, and Walker B motif in NBD are involved in nucleotide-induced conformation change in NBD region. The highly conserved aspartate/glutamate of the Walker B motif forms hydrogen bonds with the threonine/serine of the GKT/S box and with a bound water molecule and facilitates nucleotide hydrolysis (Fig. 6.20).

a ABC transporters’ various domain assemblies. The red half-circles and dashed lines indicate NBD interface, P-loops and conserved signature motifs. In all ABC transporters, coupling helices transmit conformational changes between the NBDs and the TMDs. b In single NBD, various domains like P-loop, A-loop, catalytic glutamate, LSGGQ signature sequence is shown. c Linear subunit presentation with conserved sequence
6.9.1 Mechanism of ABC Transporter
ABC transporters transport substrates against a chemical gradient, a process that requires ATP hydrolysis as a driving force. Under physiological conditions, ABC transporters operate in a single direction (either import or export) except drug efflux pump LmrA having reversible transport under acidic conditions. The transmembrane domain may face outside or inside by changing conformation. The energy for the conformational change in TMD in inward and out ward face is provided by the binding of substrate and Mg2+ ATP, followed by ATP hydrolysis and substrate release. ABC transporters have similarity to secondary transport in mechanism (Fig. 6.21).

Catalytic mechanism of ABC transporter A. The outward-facing type II importer binds to substrate at binding domain with two molecules of MgATP. The nucleotide domain dimerization because of binding results in the closed conformation with substrate confined to center of cavity mid-membrane. The ATP hydrolysis and NBD dissociation allow substrate to escape into the cytoplasm. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4338842/figure/fig-002/Page3of3
The ABC importers bind to substrate through mediator protein, while exporters directly interact with substrate through their TMD. The NBD forms catalytic domain with Walker A or P-loop, Walker B, Q-loop and H motif (switch region) and conserved sequence LSGGQ in alpha-helical region. The relative orientation of catalytic sequence to alpha-helical region is decided by ATP binding. Usually, in active ABC transporters, its subunits are arranged in head to tail in opposite direction to each other. TMD1 and TMD2 heterogeneous helices are further divided into three separate fold as Type ABC transporter, Type II ABC transporter, and ABC exporters’ folds. In outward positions, the TMD helices are in extended wing because of TM1 and TM2 from one subunit and TM3 and TM6 from other subunit. TMD primarily interacts with variable Q-loop with conserved glutamine in α-helical domain. Any change in Q-loop is directly coupled to ATP hydrolysis and conformation change of TMD. Walker B-loop participates in binding. Magnesium interaction with ATP requires D502 of Walker B. ABC transporters utilize two ATPs per one molecule of substrate transport. In addition to substrate binding, two sets of amino acids of D-, Q-, and H-loops are required for catalysis. One set acts as general base to activate water molecule to attack on γ-phosphate group of ATP, and other set of amino acids stabilizes phosphate oxygen. The amino acid E502 near Walker B, Q422 of Q-loop and H534 in H motif essentially contributes to catalysis. ATP molecule is bound at interface between TMD and NBD, and terminal γ-phosphate is positioned between P-loop and LSGGQ signature sequence. The most important factor in catalysis is the relative position between NBD and TMD domain because of ATP, ADP, and substrate binding. The transport cycles move in order of substrate binding, hydrolysis, substrate release, which induces TMD conformation change from close form with bound ATP to open form without ATP. The change in conformation stimulates substrate translocation also.
6.9.2 Classification of ABC Transporters in Mammals
ABC transporters are ubiquitously found in lung, brain, muscle, spleen, endosome, and rod photoreceptors in mammals. These transporters are classified into various classes as ABCA, B, C, D, E, F. The class ABC A transporters of 12 members are basically involved in transport of phospholipids and cholesterol to HDL. In lung, they protect cell surface (Table 6.1).
These are multidrug transporter and efflux N retinyl-diester-phosphatidyl ethanol amine. The ABC B class of 11 members are found in kidney, brain, ER, liver, and mitochondria. These are involved in multidrug resistance. They are involved in peptide transport to ER. The C class of 11 members, present in all tissues, is involved in drug resistance, organic anion, nucleoside, chloride ion transport. ABC D of four members are identified in peroxisomes. ABC E class has only one member. The ABC F is also present in all tissues. The ABC G of eight members regulates transport of cholesterol and other sterols.
6.10 Lipid Transporters in Maintaining Membrane Asymmetry
The plasma membrane has to maintain lipid asymmetry for biological functions and requires constant and regulated transport of membrane components for the purpose. The nonpolar molecules can translocate easily from inner to outer in other direction but lipids with polar group are transported by transporters in ATP-dependent manner. There are three major transporters which are known as flippases, floppases, and scramblases. The flippases are also known as P4 ATPases, which transport phospholipids to inner leaflet of membrane and have substrate specificity to phosphatidyl serine and phosphatidyl choline. The flippases’ transporters may cause change in shape of membrane like bending of the membrane for vesicle formation or vesicle budding. In erythrocyte, amino phospholipid flippases are identified for PS and PC transportation to inner membrane.
ATP8B1 flippase is a phosphatidylserine translocase in human. The other enzyme, floppases, belongs to ABC transporters’ large family, which transport polar lipids to outer membrane in energy-dependent mode for transport. P-glycoprotein floppase is a multidrug resistance protein in human.
The scramblases can transfer lipid in both the directions without requiring energy. These are activated by calcium during membrane injury, apoptosis, or coagulation and transport PS and PE to outer leaflet for destroying lipid symmetry. Phospholipid scramblases (PLSCRs) are palmitoylated proteins of lipid raft, which is under regulation by phosphorylation and dephosphorylation. Unphosphorylated form localizes in nucleus, where it interacts with topoisomerases and is required for cell division.
6.11 Aquaporins
Water is the main component of life, and its exchange is characteristic of any living being. First water channel proteins, known as aquaporins were first isolated from RBC and later from renal proximal tubule membrane Peter Agre group in 1992.
Aquaporins (AQPs), the integral proteins of membrane, are small, hydrophobic, and homotetramer proteins, which are involved in bidirectional transport. These proteins are present in all living beings. The aquaporins may facilitate the transport of urea, nitrate, NO, hydrogen peroxide, carbon dioxide (CO2), and Ammonia (NH3) along with water. Numerous identified isoforms are differentially expressed and modified by posttranslational processes for tissue-specific osmoregulation in various organisms. In mammal, aquaporins are involved in multiple physiological processes including urine concentration by kidney tubules, nerve transmission, metabolism of lipids, fluid secretion, tissue swelling, cell migration, and salivary gland secretion along with skin hydration. The genetic defects in aquaporins may lead to disorders like kidney dysfunction, loss of vision, and brain edema. According to their substrate specificity and localization, the aquaporins are divided into two subgroups:
-
1.
Classical Aquaporins are exclusively water channels.
-
2.
Aquaglyceroporins are the proteins with broader specificity for nonpolar molecules like polyols, glycerol, ammonia, CO2, NO, urea along with water and may participate in nutrient uptake and osmoregulation but bacterial glycerol facilitator GlpF only allows the movement of polyols. The unusual aquaporin 6 acts as anion channel. The aquaporins are mostly found in plasma membrane but may be localized inside the cell organelle membrane. The movement of water and other solutes through aquaglyceroporins (AQPs) supports bidirectional downhill movement through pore or channel formed by aquaporins. The number of genes identified for coding aquaporins in various organisms is variable with one or two genes in bacteria and yeasts, around 13 in mammals (7 are for Renal tubules to concentrate urine) and more than 38 genes in plants. Aquaporins are highly selective and efficient for the transport of water and glycerol but do not allow hydroxide, hydronium ion, and proton movement. This selective transport protects the membrane electrochemical gradient (Figs. 6.22 and 6.23).
Fig. 6.22 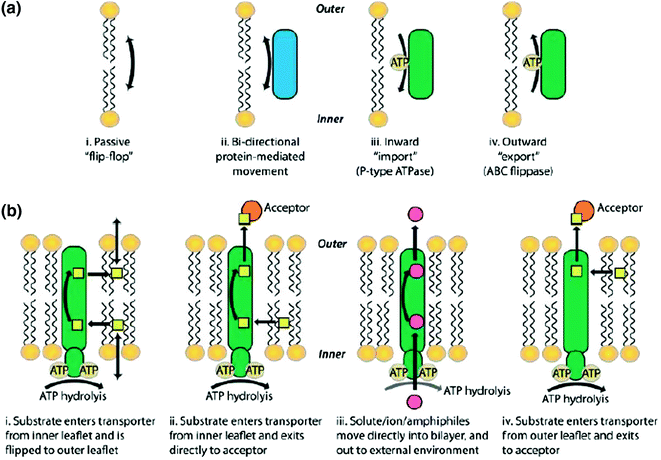
Transporters involved in maintaining lipid asymmetry in membranes. Mechanisms of transbilayer lipid movement
Fig. 6.23 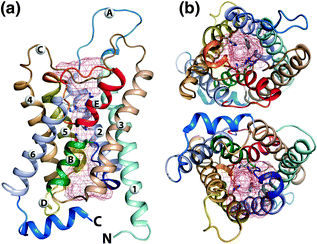
Bovine aquaporin structure. a Lateral view showing transmembrane 1–6 and connecting loops A–E with pore of red mesh. b Aquaporin from upper side and from lower side


6.11.1 Structure and Function of Aquaporins
The active form of AQPs are homo-tetramers. Each monomer of 26–30 Kd contains 300 amino acids with six transmembrane helices (TM1-6) and five connecting loops (A–E). The primary structure of aquaporin oligomer is made of symmetrical two halves, hemipore-1 (transmembrane 1–3) and hemipore-2 (transmembrane 4–6), as shown in Fig. 6.24. The transmembranes are arranged into two halves of 2, 1, 6 and 5, 4, 3, connected by loops to form channel. The four monomers together form a central pore or channel for ions and gases to move but water is not allowed through this central channel. The water diffuses through individual monomers of aquaporins. The three connecting loops A, C, E are externally located, while B and D face cytosolic side. The two loops, one from each side B and E, have signature motif of aquaporin family (major intrinsic family) asparagine, proline, and alanine (NPA). There are two conserved sequences of NPA in aquaporin channel with two asparagine side chains toward pore at the end of hemipore. All transmembranes have their N-terminal and C-terminal in cytoplasm. The B-loop entering from extracellular side and E from other cytosolic side create seventh alpha-helical domain to form a part of surface for water channel.

Aquaporin diagrammatic representations of transmembrane (1–6) and connecting loops (A–E) with signature motif Asn–Pro-Ala (NPA). a Shows the linear arrangements of the transmembrane protein showing the two separate regions of helical domains. b Two separate regions interact to form the three-dimensional orientation of the protein. The aquaporins pore is composed of two halves (hemipores). The diagram shows the two hemipores interaction to form the functional aquaporin
If water is moving from outside to inside, it has to interact with side chain carboxylic group of amino acid at the surface of aquaporin. The presence of arginine and aromatic amino acids at the narrowest part of the pore is the final selection for any molecule to enter. The overlapping of asparagine, proline, and alanine (NPA) in the center of pore reduces the size of pore for movement, which is important for water molecule interaction for selectivity. The water-specific aquaporin is observed with pore size of 2.8 Å, so bigger molecule or proton will not pass. After interaction with surface protein side carboxylic group, the water molecule will be selected by polar or nonpolar amino acid residues of pore wall proteins, which will provide channel specificity of molecule and will determine the rate of transport.
Figure 6.25 shows the various important amino acids found at pore site. The B and E short helices from opposite side of the pore with positive-charged amino acids with their side group projecting toward center play an important role in selection because of their dipole nature. At the constricted site of NPA with conserved aromatic and arginine amino acids, water ionic bonds become weak. The aromatic amino acids may vary in various aquaporins. In bovine aquaporin (AQP1), phenylalanine and arginine form constriction site. Histidine 182 at surface of pore provides ionic interaction to water molecule, while phenylalanine 58 repels water. After clearing Aro/R constriction site, the water molecule interacts with two conserved NPA sequences and its neighboring amino acids, where protruding valine pulls water molecule to interact with asparagine 194 and asparagine 78 in NPA segment. In the pore, the electrostatic interaction of the NPA repeats and the aromatic/arginine (ar/R) constriction results in the exclusion of protons. These amino acids’ segment of NPA is strong lipid anchor for changing water orientation, which is also supported by strong dipole of B and E helices to change water dipole in opposite side to previous one. The NPA amino acids are preceeded and followed by hydrophobic residues (isoleucine 193 and phenyl alanine 24 before and glycine 74, alanine 75 and valine 181 after NPA sequence). These hydrophobic residues stop water interaction with other amino acids of wall proteins for faster movement. Tyrosine 31 of D-loop is found responsible for closing of channel because of its insertion into NPA site. The phosphorylation at serine 107 of aquaporin induces change in conformation, which may open aquaporin channels, because serine 107 is present in B-loop, which is directly connected to tyrosine 31. These channels are very fast to have permeability of water 3 × 109 mol/s.

Bovine aquaporin (AQP1) detail structure of pore region with conserved NPA provides interaction mechanism of channel with water. Half helices dipoles and the hydrophilic and hydrophobic residues lining the pore are shown in red and yellow, respectively; the aromatic/R and NPA selective filters are shown as well
The aquaporin channels may be regulated by concentration gradient of water, phosphorylation and dephosphorylation, temperature, and pH. This has been observed in plants that decrease in pH may close the channel because of histidine protonation in D-loop. The mercury chloride binding to cysteine at NPA site also closes aquaporins channel. Vasopressin in kidney may activate channels through cAMP-induced phosphorylation. The genetic defect in human aquaporins AQP2 leads to diabetes insipidus. Ca 2+ is found to regulate translocation of AQP2 to the plasma membrane. AQP2 overexpression because of heart failure leads to water retention. AQPs role in homeostasis of the cerebrospinal fluid (CSF) and neuron excitability is well-established (Table 6.2).
6.12 Active Transport Through Group Translocation in Bacteria
6.12.1 Phosphoenolpyruvate (PEP): Carbohydrate Phosphotransferase System
The bacteria take nutrients from their surroundings by various modes of transport but the transport of sugars and its derivatives like sugar alcohols, amino sugars, glucuronic acids, disaccharides is transported by group translocation with histidine heat resistance protein HPr as high-energy phosphate donner protein. The transporting sugar molecule is also phosphorylated during transport, which makes sugar negatively charged. The phosphorylation of sugar does not change intracellular concentration gradient and also check sugar leakage from cell. The seven families of phosphotransferases have been identified in various groups of bacteria with mostly conserved sequences and dissimilarity in substrate specificity. During this transport, the sugar is chemically modified across the membrane and that’s why this transport is also called as group translocation transport. The phosphortransferases complex has cytoplasmic, peripheral, and integral proteins. The cytoplasmic proteins are constitutive and nonspecific, which are involved in group translocation of various other proteins also. The cytoplasmic proteins comprise of heat-resistant histidine protein HPr and Enzyme E1. E1 protein has histidine 189 at N-terminal, which participates in reversible phosphorylation of HPr, and its C-terminal domain is required for protein autophosphorylation by PEP. HPr is small monomeric protein, which is phosphorylated at N-terminal histidine 15 by E~P. The cytoplasmic PTS proteins or membrane-associated hydrophilic PTS domains undergo transient phosphorylation during transport of sugars. The integral membrane proteins are known as Enzyme II (EII), which participate in substrate translocation and its phosphorylation. The EII enzymes are inducible and substrate-specific proteins, which include EIIA, cytoplasmic protein, A peripheral protein associated with inner membrane EIIB, and carrier integral protein EIIC. EIIA and EIIB are sugar-specific proteins. EIIA for glucose is phosphorylated at histidine 90. The EII component may be separate or fused in different organisms. EIIA is conserved; EIIB is hydrophobic which is phosphorylated at conserved cysteine residue by EIIA. EIIC has 6–8 transmembrane regions with one conserved GXXE motif in hydrophobic loop. Protein EIIC has three periplasmic (2, 4, 6 loops) and two cytoplasmic (3 and % loops). In Bacillus subtilis, oligo-β-glucoside-specific PTS has three separate enzymes, while in the same organism, mannitol PTS has EIIB fused with EIIC.
The PTS catalyzes the general reaction of sugar transport as
The reaction cycle starts with transfer of phosphate group from PEP to a histidine residue on Enzyme I. The Enzyme EI transfers this phosphate to HPr protein on histidine 15. In the next step, this phosphate group is transferred from HPr to EIIA protein again on histidine residue. The specific Enzyme EIIA for sugar transfers phosphate to the cysteine residue of Enzyme IIB. Finally, Enzyme EIIB transfers phosphate group to EIIC bound sugar, which is phosphorylated and translocated to cytoplasm (Fig. 6.26).

Transport of sugar fructose (PTS), mannitol (PTS), and cellobiose (PTS) by group translocation in Bacillus subtilis, which has nine complete PTS, six PTSs without EIIA component, and one PTS without EIIA and EIIB
The majority of sugars are transported in phosphorylated form but fucosyl-α-1,3-N-acetylglucosamine in Lactobacillus casei is translocated in unphosphorylated form exceptionally. Some sugars are immediately dephosphorylated after translocation in cytoplasm like mannose in Enterobacter faecalis. The glucose uptake is enhanced by the availability of nitrogen in bacteria through these phosphotransferases.
6.13 Light-Driven Transport
The various organisms have specialized proteins of rhodopsin family to seize light energy for various physiological functions. The rhodopsin protein part, opsin, is covalently linked to retinal chromophore, which may be in trans- or cis-configuration. The animal rhodopsin (also known as Type II) are cell G-coupled receptors, while microbial rhodopsin (Type I) may act as ion pump, ion channel, sensor, photosensory receptor, regulator for gene expression or a kinase. Halobacterium salinarum has normal respiration in the presence of O2 and high nutrients, but in the absence of nutrients, it survives by using light energy, which is identified with purple patch on the bacterial membrane because of retinal base. The three microbial pumps are well-characterized in their structure and function known as the bacterio rhodopsin H+ proton pump, channel rhodopsin for chloride ion. The Halobacterium salinarum has sensory rhodopsin I and sensory rhodopsin II, which activate transducers for senses (Fig. 6.27).

Classical four microbial rhodopsins from the archaeon Halobacterium salinarum. The membrane of H. salinarum contains four rhodopsins, bacteriorhodopsin (BR), halorhodopsin (HR), sensory rhodopsin I (SRI), and sensory rhodopsin II (SRII, also called phoborhodopsin, pR). BR and HR work as a light-driven
The animal and microbial rhodopsins are transmembrane of 7 α-helices, covalently linked to retinal through lysine residue and form protonated Schiff base with it. The retinal chromophore may be in cis- or transconfiguration. The ground state of microbial and animal rhodopsins has all trans- and 11-cis-retinal configuration (Fig. 6.28).

Synthesis of retinal chromophore in microorganism and animal from β carotene. The ground state of microbial and animal rhodopsins possesses all-trans- and 11-cis-retinal as its chromophore, respectively, bound to a Lys residue via a Schiff base, which is normally protonated and exists in the 15-anti configuration. It should be noted that microbial rhodopsins depend exclusively on all trans-retinal, while some animal rhodopsins possess vitamin A2 (C3=C4 double bond for fish visual pigments) and hydroxyl (C3–OH for insect visual pigments) forms of 11-cis-retinal. Usually, photoactivation isomerizes microbial rhodopsin selectively at the C13=C14 double bond and animal rhodopsin at the C11=C12 double bond. Post-content
Thermodynamically, retinal is preferred in all transconfigurations in nonpolar environment with absorption maxima at 360 nm wavelength but the protein interaction with retinal shifts this absorption maxima to broader range in visible light range from 400 to 600 nm. The light absorption by retinal stimulates isomerization from trans-cis to cis-trans in less than a fraction of second. The energy released by this transition isomerization induces change in opsin protein conformation for various activities. The sequences of animal and microbial proteins are quite dissimilar in spite of both having seven transmembranes. Bacteriorhodopsin translocates one proton inside to outside in cyclic reaction and generates membrane potential across the membrane for ATP synthesis. The channel rhodopsin of H. salirium transports chloride ion from outside to inside of cell for ATP synthesis and for osmotic balance. The sensory rhodopsin I (SRI) acts as stimulator of positive phototaxis, while SRII works oppositely. The bacterial movement toward particular wavelength light is positive phototaxis and moving away is negative. SRI (580 nm \(\lambda_{\hbox{max} }\)) and SRII (500 \(\lambda_{\hbox{max} }\)) perform through their transducers and cotransducers. The cotransducers are named as HtrI for SRI and HtrII for (SRII). SR proteins with their cotransducers are localized in cell membrane in 1:1 ratio to form tetramer. The signal induced by retinal trans-cis isomerization by light is propagated to cytoplasmic transducers CheA and CheY resulting in phosphorylation and DEphosphorylation for flagellar movement in desired direction (Fig. 6.29).

Bacteriorhodopsin molecule is purple and is most efficient at absorbing green light (wavelength 500–650 nm, with the absorption maximum at 568 nm). Bacteriorhodopsin has a broad excitation spectrum
The crystallographic studies and electron density of catalytic side have explained the photocycle of ion transport. The seven transmembranes of opsin protein may be designated as A B C D E F G, which are arranged to cover centrally linked protonated retinal Schiff base to G-helix by lysine 216. Basically, the light absorption by retinal redistributes its own electrons in protonated base resulting in isomerization, proton transfer between various amino acids at catalytic side, water relocalization, and finally change in protein conformation for physiological function. The water molecule at 402 position in catalytic site accepts proton from retinal and transports it to aspartic acid 85 and asp21. The protonation of Asp85 and retinal deprotonation induce proton movement to outside of membrane, and proton is released. Cytoplasmic proton migration protonates retinal Schiff base to its original position to complete light-induced cycle. The conserved residues involved in proton transfer are shown in Figs. 6.30 and 6.31.

a Structure of bacteriorhodopsin (BR), with conserved aromatic residues highlighted (PDB ID: 1QM8). Tyr83, Trp86, and Trp182 are strongly conserved among microbial rhodopsins (orange). Aromatic amino acids are strongly conserved at the position of Tyr185, Trp189, and Phe219 (yellow). In BR, Trp86, Trp182, Tyr185, and Trp189 constitute the chromophore binding pocket for all transretinal configuration (gray). b Crystallographically observed internal water molecules of BR (shown as green spheres)

a Mechanism of proton transfer by bacteriorhodopsin. The rhodopsin active site detail by electron density map with all transretinal, W86 and W182 covering polyene chain between 9 and 13 methyl group to immobilize retinal site. The H-bond formation from Schiff base to extracellular surface through water 402, D85 and water 401 is visible. Water 501 links A215 carboxylic group to W185 indole nitrogen, b R82 in extracellular half-channel, where its guanidinium group interact with Schiff base through water 406, 401, and D85. Water 407 stabilizes positive charge. Asp85 and Asp212
At the ground state of stable active site, the positively charged retinal Schiff base interaction with three water molecules 401, 402, 406 and aspartic acid (D)85, D212 stabilize separate noninduced light changes fat active site. The H-bond interaction of aspartic acid D85 with threonine T89 asp D212, tyrosine Y57, Y185, and water molecule 406 keeps Asp D85 in anionic form. The aspartic acid D85 and water 406 also interact with positively charged arginine R82. The transition of retinal base from trans to cis by light will dramatically destabilize this ground state. The hydrogen bond between these amino acids and water will be disturbed and rearranged, which will cause to disassociation of proton from Schiff base. The asp D85 is first proton acceptor which is centrally located. Water 402 actively participate in hydrogen bond rearrangement because of its position its H+ and OH− easily may protonate and deprotonate neighboring residues. The extracellular side of retinal linked to Arginine R82 interacts with three-dimensional hydrogen bond mesh contributed by water molecule 404, 405, 406, and 407 and amino acids. The retinal molecule is linearly connected to arginine 82 through H bonds starting from water 402-D85-water, 412-Arg R82, and other water 402–D212-W57-water407-arg R82NH21/NH22. The whole mesh is interconnected.
The proton release complex with water 403, 404, 405, Glu E 194, Glu E 204 is separated by isoleucine I78 and leuL201 from hydrophilic environment. At the C-terminal of D helices transmembrane, a unpaired buried arginine R134 interacts with salt bridge to glutamate E194 and interacts with carboxylic group of amino acids at 126, 128, and 194 positions. The transmembrane G at cytoplasmic side gets kink because of π bulging at alanine A215, which results in peptide bending between alanine A 215 and lysine K216. This bending stimulates local rearrangement of H bonding between water and amino acids (Fig. 6.32).

Proton transfer interaction of bacteriorhodopsin retinal Schiff base to various amino acids of opsin protein. Representation of retinal Schiff base catalytic domain by electron density map, depicting protonated retinal base and water 402 tightly hydrogen bonded between positive charge Schiff base and two anions D85 and D212. The Schiff base nitrogen atom, water 402, and the two acceptor oxygen atoms of Asp85 and Asp212 are in single plane
The new hydrogen bonding between molecules and new local arrangements around G transmembrane helices slightly tilt this from canter, resulting in C-terminal displacement to outer side, and induce change in conformation. The water molecule at 502 stabilizes this change through interactions with amino acids and carboxylic group of retinal Schiff base. The rearrangement of G helices and water molecule amino acids interactions decrease PKa of aspartic acid D96, which donates proton in photocycle and control reprotonation of aspartic 96 by isomerization of retinal Schiff base from cis to all trans (Fig. 6.33).

In bacteriorhodopsin helices G at cytoplasmic side, A kink between alanine A215 and lysK216 is produced because of alanine 215 π-bulging. The local hydrogen bonds between water and amino acids are rearranged to release proton
6.14 Pore-Forming Toxins
Pore-forming toxins (PFTs), the bacterial virulence factor, single major family of proteins, are secreted by gram-positive and gram-negative bacteria. PFTs may be present either in cytoplasm or in the membrane. The conformational change in cytoplasmic PFTs may translocate them to membrane in need of hour. The numbers of bacteria like Staphylococcus aureus, E. faecalis, E. coli, and Vibrio cholera had shown the presence of these proteins during pathogenesis. The size of pore may be small (0.5–4 nm) or large (more than 20 nm). Mostly, PFTs are water-soluble. Binding of pore-forming toxin to host cell receptor through sugar, lipids, or protein leads to change in conformation for pore formation. The strength of pore formation is directly proportional to their virulence power. As per transmembrane structure of PFTs in host cell membrane, the PFTs are divided into two groups as α-PFTs because of α-helical pore and β-PFTs because of β-barrel pore (Fig. 6.34).

Structure of pore-forming toxin a single peptide and b multimeric proteins Proteins Data Bank ([PDB] accession number 1QOY). c Structure of aerolysin, a β-PFT produced by A. hydrophila (PDB accession number 1PRE). d PFO monomer (PDB accession number 1PFO). e Speculated arrangement of CDC monomers into multimeric pore by mapping PFO monomers onto a PLY cryo-electron microscopy (cryo-EM) image (PDB accession number 2BK1). Structures were visualized using PyMOL (d) or MMDB (486) (a–c and e)
The various molecules in the host membrane may act as receptor molecules like glycosyl phosphatidyl inositol (GPI), chemokine receptor (CCR5 or CD19) of white blood cells, disintegrin and metallo-protease (ADAM)-type proteins, cholesterol, and other lipids. Pore-forming toxins may change their conformation to adjust in host environment. The pore formed by PFTs may be continuous barrel layer of proteins or may be in toroidal form because of lipid protein interaction.
The major pathogens producing different PFTs proteins are shown in Table 6.3.
The PSTs act first by binding to receptors, which are followed by oligomerization, change in conformation, and insertion into host membrane to form pore. Till date, six families of pore-forming toxins, including three α-PFTs and three β-PFTs, are known. α-pore-forming toxins are heterogeneous group with various PFTs like cytolysin of E. coli, exotoxin of Pseudomonas aeruginosa, and diphtheria toxin from Corynebacterium diphtheria.
The various PFTs may form various types of pores with different characteristics. The E. coli produced α-PFTs colicins family produces pore in other bacterial inner membranes. The isolated colicin A and E1 PFT, according to their function, may be divided into three domains for receptor binding, translocation, and pore formation. The pore-forming domain of colicin has ten alpha-helices. Two of these hydrophobic helices are surrounded by eight amphipathic alpha-helices. The hydrophobic α-helices are inserted into inner membrane to make nonspecific voltage-gated ion channel. This pore destabilizes membrane potential of host cell. The other αPFT such as insecticidal cry toxin from Bacillus thuringiensis, diphtheria toxin translocation domain, and eukaryotic protein Bak, has shown similar structure like colicin for pore formation. The colicin PFT of this group forms the pore by binding to receptor, translocates to periplasm, and causes cell death by forming pore in inner membrane. Bcl-2 completes pore formation in mitochondria by releasing cytochrome C to activate caspases for apoptosis.
A member of αPFT cytolysin family, cytolysin A(Cly), hemolysin E(Nhe) enterotoxin of Salmonella enterica and Shigella flexneri and E. coli forms pore of 2–7 nm diameter for cation in macrophage leading to apoptosis. Cytolysin A is found to be α-helical protein in elongated shape with short hydrophobic β-strands in tongue shape (βtongue). During formation after assembly, the β-strands become disassociated from the protein for insertion into lipid bilayer; this event causes the rearrangement of N-terminal region of amphipathic helices at the pore site. The N-terminal domain arrangement may vary in length but this variation cause is not clear till now. The conformational change by the N-terminal amphipathic alpha-helices catalyzes a very organized step-by-step oligomerization of monomers.
There are various ways of protein–protein or lipid–protein interaction for pore formation as shown in Fig. 6.35.

α-PFPs may form pores in various ways (a). Protein-lined pores, b protein–lipid pores, c protein–lipid pores: hybrid pores, d structure of the pore formed by FraC and liposomes (PDB: 4TSY). The bridging lipids connecting two adjacent molecules of FraC are shown in the bottom left side (green)
When toxin protein interacts with host lipid for pore formation, lipid may provide a curved nonbilayer shape at pore periphery. In this pore, the protein regulates stress caused by distortion of membrane. As seen in eukaryotic β-barrel actinoporin PFT from sea anemones, in which cation selective pore is formed by interaction with sphingomyelin of host membrane. Usually, the toxins recognize and interact with specific lipid domain of sphingomyelin clustering or cholesterol–sphingomyelin complexes, which support the accumulation of PFTs locally to ease their function of pore formation. This may result in lamellar structure destabilization by lipid transmovement across the membrane (Fig. 6.36).

βPFTs, hemolysin, aerolysis, and CDC family pore formation by extraction and insertion and interaction with other monomers of prestem loop of monomers to form beta-barrels
The β-pore-forming toxins include aerolysin and cholesterol-dependent cytolysin family (CDC), and these are secreted majorly by gram-positive bacteria and minor by gram-negative bacteria. Gram-negative Aeromonas SAPs produces aerolysin as protoxin. The C-terminal cleavage converts protoxin into toxin protein, which is elongated multidomain protein. The aerolysin protein may be divided into four domains; the domains 1, 2, and 4 are together, and domain 3 is separated. The domain three has multiple short α helices with β-sheet. The domain four (4) interacts with cholesterol for binding by inserting several loops. The domain four (4) has conserved domain of tryptophan residues with three short hydrophobic regions known as L1, L2, and L3. The domain 3 has two transmembrane α-helices in soluble form, which are converted to β-hairpin transmembrane during pore formation.
While α-PFTs start pore formation by the insertion of hydrophobic helices, the β-barrel formation occurs in concerted mode (Fig. 6.37).

Actinoporin and colicin family PFTs pore formation by monomer alpha-helical hydrophobic segment extraction and insertion into host membrane, umbrella formation, oligomerisation either into two or more peptides
As mentioned above, the other major family of β-pore-forming toxins is cholesterol-dependent cytolysins (CDC), which include perfringolysin O (PFO) of C. perfringens, streptolysin O (SLO) of Streptococcus pyogenes, pneumolysin of S. pneumoniae, and listeriolysin O (LLO) of Listeria monocytogenes belong to cholesterol-dependent cytolysin family. These toxins are produced mostly by gram-positive bacteria and by few gram-negative bacteria. The pore formation is dependent on cholesterol for pore formation. In cholesterol-dependent cytolysin toxin, pore may contain from 20 to 50 proteins subunits, and each subunit donates 2 β-hairpin for pore unlike other β-PFTs. Cytolysins are produced as monomer or dimer and have some similar features like eukaryotic membrane attack complex (MAC). Cytolysin also has four domains like aerolysin in elongated structure and makes well-defined prepore in the membrane. The CDC, unlike some other toxins, which rearranges their structure for pore formation, goes under transition from alpha-helices to their β hair pin secondary structure at membrane surface (prepore).
The transmembrane interacts with lipid cholesterol and forms arc-like structure to create Gap (semi pore) in the membrane. At this stage, oligomerization cytolysin monomers occur. The oligomerization transforms the surrounding arrangements of molecules to form functional pore. The conserved tryptophan in toxin and cholesterol play an important role in pore formation (Fig. 6.38).

Depiction of pore formation pathway of pore-forming toxins (PFTs). Soluble PFTs are recruited to the host membrane by protein receptors and/or specific interactions with lipids (e.g., sphingomyelin for actinoporins or sterols for cholesterol-dependent cytolysins (CDCs)). Upon membrane binding, the toxins concentrate and start the oligomerization process, which usually follows one of two pathways. In the pathway followed by most β-PFTs, oligomerization occurs at the membrane surface, producing an intermediate structure known as a prepore (mechanism 1), which eventually undergoes conformational rearrangements that lead to concerted membrane insertion. In the pathway followed by most α-PFTs, PFT insertion into the membrane occurs concomitantly with a sequential oligomerization mechanism, which can lead to the formation of either a partially formed, but active, pore (mechanism 2)
α-hemolysin (Hla) of cholesterol-dependent cytolysin (CDC) family anchors through N-linked glycan proteins of hosts. Aerolysin binds through glycosylphosphatidylinositol (GPI) anchors. Colicins f α-PFT family interact with lipopolysaccharides (LPS) in the outer bacterial membrane. α-PFT fragaceatoxin C (FraC) binds to sphingomyelin clustering.
The eukaryotic cell also has pore-forming toxins as the mechanism of their defense, which is known as membrane attack complex (MAC) or performin toxins. The T cell secretes them to destroy pathogen. These toxins like CDC have two transmembrane domains. The hydrolysin from cnidaria (phylum of aquatic invertebrate animals), enterolobin toxin from Enterolobium contortisiliquum, and human C8 are the examples of eukaryotic toxins.
6.14.1 Activated Signal Pathway and Toxin Effect in Host Cells
The various defense mechanisms against these virulent pore-forming toxins exit in host like human. The binding of toxin to cell membrane may also activate p38 mitogen-activated phosphokinase (MAPK), c-JUN N-terminal kinase like MAPK-KGB1 pathway. The detail of receptor signaling is discussed in Chap. 9.
PFTs pore formation may result sometimes in increase in extracellular potassium as a defense mechanism. The potassium efflux activates nucleotide-binding oligomerization domain (Nod)-like receptors (NLRs), which have variable N-terminal domains such as caspase recruitment domain (CARD) and pyrin domain (PYD), which activate apoptosis and inflammation pathways. These receptors are found on immune cells like lymphocytes, macrophages, dendritic cells, and also in nonimmune cells and play a role in innate immunity, phagocytosis, and killing of pathogens. The caspase 1 induces sterol regulatory element binding protein (SREBPs), the regulator of membrane lipid synthesis. The extracellular potassium increases in response to toxin interaction and also activates caspase 2 for inducing apoptosis. The activation of p38 and extracellular response-activated kinase (ERK) brings back potassium level to normal. α-defensin proteins are also detected in host to act against PFTs (Fig. 6.39).

Source Cosentino et al. (2015)
Effect of pore-forming toxin in vivo on host cell by various path ways.
Calcium efflux in response to PFTs activates the release of lysosomal enzymes such as sphingomyelinase and hydrolase to degrade pathogenic molecules Endocytosis is another defense used by host cell. PFTs also interact with cellular signaling, and molecules such as arthritis were caused by γ-hemolysin in human. The hemolysin induced the expression of interleukin 6 in osteoclasts. The downregulation of interleukin 12 and nitric oxide synthetase is observed in macrophage by cytolysin toxin.
6.15 Ionophores
Ionophores are natural and synthetic organic molecules of diverse type, which can transport cations by forming lipid ion-soluble complex. Each ionophore possesses a unique property of changing transmembrane ion gradient and electrical potential, synthesized by bacteria, and acts as protector against competing microbes. They disrupt membrane’s ionic gradient required for proper transport; therefore, they have got antibiotic properties. Ionophores increase the permeability of biological membranes to certain ions and antibiotics; that’s why they are used as growth enhancers as additives in certain animal feed. The various ionophore antibiotics are macrolides. The two classes of ionophores are identified as carriers and channels.
The carriers bind to specific ion, shield it from hydrophobic environment, leave the ion on other side of membrane by diffusing across the membrane, and come back without ion to original place. The carrier polyether ionophores include approximately more than 130 types having antibiotic properties against bacteria, fungi, and virus. Some of them are found to have antitumor activity also. The antibiotic property of these polyether ionophores structure specifies their cation metal binding and is directly correlated to their antibiotic property. The carrier ionophores are not found to be effective in gram-negative bacteria because of their cell wall but the opposite is true for gram-positive bacteria. The gram-positive bacterial membrane is permeable for these small molecules. Because of this reason, E. coli is found to be important for food and human health. The carrier ionophore examples include valinomycin, calcimycin, salinomycin, monensin. Valinomycin is produced by various strains of Streptomyces species as S. tsusimaensis and S. fulvissimus. The valinomycin is highly selective for potassium and Rubidium (Rb+) ion and does not allow sodium or lithium transport (Fig. 6.40).

Valinomycin-A carrier ionophore, A, dodecapepsipeptide ring structure, B, L valine-d hydroxy isovaleric acid and D val-L lactic acid, unit arrangement
The binding and affinity constant for the potassium–valinomycin complex are ten times higher than sodium. Valinomycin was observed to downregulate the lymphocytes multiplication induced by phytohemagglutinins in human. Valinomycin–metal ion interaction essentially requires dehydrating form of ions, and complex is formed in energy-dependent process. The amount of energy required is the deciding factor for forming valinomycin–potassium and rubidium complex but not with sodium (Table 6.4).
As shown in Table 6.3, because of unequal distribution of ions across the membrane, the electrical potential across the transmembrane is generated, which initiates binding of metal ion to ionophore for diffusion. The movement of ions may decrease or balance electrical potential of membrane. The ionophores may reduce membrane electrical potential through ions movement, which is the basis of their antibiotic property. These metal ion ionophores act as uncouplers like the cell’s own uncouplers in mitochondrial respiration. The synthesis of ATP becomes disassociated from electron transport chain. Valinomycin acting as antibiotic may kill the cell by disturbing electrical gradient in transport and energy-generating process. In S. aureus, Listeria innocua, L. monocytogenes, B. subtilis, and Bacillus cereus, gram-positive bacteria the Valinomycin activity is pH dependent. Recently, the valinomycin is reported to inhibit severe acute respiratory-syndrome corona virus survival at 3.3–10 μM (Table 6.5).
The structural study of carrier ionophore by X-ray has revealed the binding site of ions through oxygen atoms in the center of ionophore. Ionophores are neutral complexes with monovalent cations (monensin, salinomycin) and with divalent metal cations (lasalocid, calcimycin). The carboxylic group is deprotonated at physiological pH, so more of neutral complexes are found in the cell. The pseudocyclic structure formed by cation may be stabilized by intramolecular hydrogen bonds of carboxylic group and hydroxyl groups.
The hydrophobic residues of ionophore encircle the ionic interactions for the protection. The polyether ionophore anion (I–COO−) binds the metal (M+) cation or proton (H+) and forms either (I–COO−M+) or (I–COOH) depending on pH. This interaction changes Na+/K+ ATPases activity to change in gradient of the cell. Resultantly, this may cause the death of the bacteria cell because of increase in osmatic pressure, swelling and bursting of cell by disturbed ATPases. The coordinated geometry between ionophores and ions is important for its function.
The other examples of carrier ionophore, isolated from Streptomyces chartreuses, are calcimycin (A23187) and ionomycin calcium ionophore. Ionomycin hydrolyzes phosphatidyl inositides in the T-cell membrane to activate phosphokinase C (PKC) through autophosphorylation by phosphorylating T helper cell (CD4) and T suppressor (CD8) cells. The hemisodium, synthetic ionophore, transports sodium ions.
The calcium ionophore calcimycin, oxidative phosphorylation, and electron transport chain uncoupler are often used for enhancing thyroid and insulin secretion. Calcimycin, because of fluorescent property, may be used as biomarker in membranes for transport study (Fig. 6.41).

Crystal structure of 2:1 complex of calcimycin with the magnesium cation. https://www.hindawi.com/journals/bmri/2013/162513/fig6/
Calcium ionophores promote three types of signal in calcium-resistant cell for cytosolic calcium increase: (1) Activate receptor-mediated calcium transport into cytosol by native Ca2+ channels, (2) Phospholipase C-dependent calcium mobilization from intracellular stores like ER, (3) At low concentration of Ca2+, ionomycin in endothelial cell, like histamine, increases cytosolic calcium, while in fibroblast, only intracellular calcium from organelle store releases out. Beauvericin ionophore has both antibiotic and insecticidal property. This has phenylalanine and hydroxyl-iso-valine amino acids alternatively to form cyclic hexodepsipeptide. It transports alkaline and alkali metals across the cell membrane. Beauvericin, found in Beauveria bassiana, wheat, barley, and corn, causes apoptosis in mammals additionally. The other class of enniatins ionophores are found in fusarium fungi. Usually, proteins have the characteristic of forming channels in the membrane for transport of various molecules, but some of the ionophore also transport by forming a channel in the membrane. These ionophores have a few unusual amino acids normally not found in proteins. Gramicidin, isolated from Bacillus brevis, has 15 amino acids, linearly arranged in D and L alternate fashion with amino-terminal formaldehyde and carboxyl terminal ethanol amine. The hydrophobic amino acids in gramicidin support its function in channel formation. These channels have outer lining of hydrophobic amino acids to interact with lipid bilayer of membrane with hydrophilic interior for ion interaction. These channels allow very fast diffusion of ions through them in very short period unlike carrier ions. Their size is larger than protein channel. Gramicidin channels allow protons and other cation movements but not of divalent cations like Ca 2, which block them. Gramicidin has antibiotic property against gram-negative bacteria, gram-positive bacteria, and pathogenic fungi but sometimes its hemolytic activity may limit its use. In various solvents, gramicidin has stable amphiphilic antiparallel β-sheet structure with a polar and a nonpolar surface as α-helix is not possible because of alternate d- and l-amino acid residues. Gramicidin has 6.3 residues per turn in β-sheet structure with channel diameter approximately 0.4 nm. The ethanol residue is projected on the surface and formyl group inserted in membrane. The nonpolar amino acids play a role in microbial specificity, and disrupted beta-sheet structure adds to its antimicrobial activity (Fig. 6.42).

a Structure of beauvericin. b Structure of calcimycin and c Nigericin
The natural mixture of gramicidin, known as gramicidin A, is observed fluorescent because of tryptophan amino acid. Its antimicrobial activity may be through gene regulation as E. coli RNA polymerase was found to be inhibited by gramicidin (Fig. 6.43).

Structure of gramicidin
Gramicidin is not synthesized by ribosomes but by cellular enzymes, and at 11 position, it has tryptophan which is replaced by phenylalanine in gramicidin B and tyrosine in gramicidin C, respectively. The interior of the channel is formed by the polar amino acids as discussed above. Gramicidin has affinity for ions in the following order as maximum for cesium and minimum for lithium Cs+ > Rb+ > K+ > Na+ > Li+.
Gramicidin is also used to stimulate the production of cytokines interferon. The property of ionophore to induce apoptosis in cancer cells is being used for drug targeting especially sodium and calcium, because ionophore has capacity to disrupt lysosomal function and can inhibit proteasome function for apoptosis (Fig. 6.44).

Source Kelkar and Chattopadhay (2007)
a Top view of the gramicidin channel as a space-filling model (color code: white, carbon atoms; blue, nitrogen atoms; red, oxygen atoms) made using RASMOL ver. 2.7.2.1 and using coordinates from PDB 1MAG. Note how the alternating l–d arrangement allows all amino acid side chains to project outward from the channel lumen and the channel lumen is lined by the peptide backbone. b Structure of the selectivity filter (residues 75–79) of the KcsA K+ channel. Two subunits are shown in ball-and-stick representation (made as in (a), color code: same as in (a)) using coordinates from PDB 1BL8. Note that all amino acid side chains extend outward from the selectivity filter.
6.16 Porins in Biological Membranes
The bacterial and eukaryotic membranes are found to have special proteins in their outer membrane for transport, known as porins which form channels for various small size solutes, ions, and other nutrients. The porins found in mitochondria are also known as voltage-dependent anion channel (VDAC). The mitochondrial conserved porin proteins are tissue specific, but in bacteria, these porins constitute a diverse group because of bacterial variable environment. The porins are made of 14, 16, 18, or 19 β-barrel strands in slightly tilted form. The porins may be monomer but mostly are trimers. Based on their structure and function, porins are broadly classified as 16-stranded nonspecific β-barrel such as Omp32, OprP, and PhoE, 18-stranded specific β-barrel (LamB from E. coli), 14 β-strands and monomeric such as OmpG and multidomain like CymA from bacteria. T. thermophilus which is P100 porin protein with N-terminal peptidoglycan binding domain and a coiled coil domain with large pore size. NMR and crystallographic studies have revealed mitochondrial VDAC pore size of 4.5 nm height and 3 nm diameter. The VDAC has 19 β-strands with N-terminal helix buried in pore.

Three-dimensional structure of the porin OmpF from Escherichia coli. Surface and internal loops are shown in dark gray. The extracellular space is located at the top of the figure, and the periplasmic space is at the bottom. The trimer is shown
The sugar-specific porins (Fig. 6.45) may allow solute up to 800 Kd. The bacterial porins are oval in shape with pore size 3–3.5 and 5 nm height. The porin first and last β-strands are joined in antiparallel mode to form complete pore.
These β-strands are connected to each other from cytoplasmic side by 8–9 long loops and 7–8 small turns from periplasmic side. The loop 3 from the cytosolic side in all porins is folded in side in barrel as shown in Fig. 6.46 and regulates pore size. Usually, loop 3 is found between 5 and 6 strands of barrel. This has mostly polar amino acids aspartic acid and glutamic acids. The L1, L2, and L4 interact with each other monomer to form trimer. L5, L6, and L7 are present on the surface of porin. The loop L8 being folded into interior contributes in channel opening toward extracellular side (Fig. 6.47).

Omp32 porin of E. coli from upper side (left-hand side) and lateral view (right-hand side) in colors with the N terminus, blue, and the C terminus, red. The longest loop L3 is labeled, as well as the additional loops (L4–L8), turns (T1–T6), and β-strands (β5–β13) structures

Mitochondrial porins mVDAC, lateral and upper view (PDB code: 3EMN) in ribbon depiction. Color—blue (N terminus), red (C terminus) with approximate molecular dimensions. The first and last β-strands (β1 and β19) are paired in a parallel way (yellow arrows). The upper view highlights N-terminal α-helix localized inside the barrel
The aromatic amino acids like tyrosine and phenylalanine are predominantly present in inner side and outer side of pore. The tyrosine is more toward outer side and phenylalanine to cytosolic side. All porins are found to have phenylalanine at their C-terminal which is required for proper folding and import. The algE porin from P. aeruginosa (18 β-strands) has positive amino acids like arginine inside the pore to translocate negatively charged alginate polymer.
The mitochondrial porins have three isoforms and are of round shape. The first and last β-strands of 19 strands pair parallel teach other unlike bacterial porins. All strands are connected through short loops, little elongated to cytoplasmic side (2–7 number) (Fig. 6.48).

Mitochondrial porins: Summary of MOM structures of β-barrel topology. Summary of the two principle architectures of MOM proteins of β-barrel topology shown embedded in the MOM lipid bilayer. VDAC and Tom40 belong to the protein families comprised by a 19-stranded β-barrel topology. Sam50 is the only representative of the MOM based on the 16-stranded barrel architecture. Functions assigned to the individual classes of proteins as long as they are clearly confirmed and marked accordingly. All structure figures are prepared using the PYMOL program
The mitochondrial porins are imported by TOM protein and inserted by sorting and assembly machinery (SAM complex) Bacterial porins are inserted by β-barrel assembly machinery (BAM) complex. In 16-stranded β-barrel porins of nonspecific class, the transport depends on molecular mass and rate of transport is directly proportional to concentration gradient. The specific 18 β-barrel porins like lamb maltoporin of E. coli have specific binding site for substrate inside channel and shows Michaelis–Menten kinetics of transport. Many prokaryotic porins are closed at high voltage of 100–200 mV. VDAC also has voltage dependence and can be easily gated by change in pH, and their permeability is controlled by membrane potential. They are involved in transport of ATP and ADP; that’s why a large number of positively charged amino acids are present in interior wall of pore.
6.17 Transport by Channel Proteins
The protein ion channels are passive transporters, are made up of transmembrane, are selective for their substrate ions, and have much faster rate of transport than pumps. These channel proteins do not interact with transporting ions and have two conformations closed and open, to regulate flow of ions. The channels may be classified into ligand-gated channel and voltage-gated channel. Ligand-gated channel opening and closing depends on the interaction with positive or negative ligand. The binding of ligand induces conformational change in protein for opening or closing. On structural basis, ligand channel (LGIC) is classified into three classes.
-
1.
Cysteine loop receptors (Nicotinic Ach) superfamily, which has pentamer cysteine loop such as nicotine acetyl receptor 5 hydroxyl tryptamine, Zn-activated channel cation transport, gama-aminobutyrate (GABAA), strychnine-sensitive glycine receptors for anions, glutamate-gated chloride channel of C. elegans.
-
2.
Tetrameric ionotropic glutamate receptors, which has four subunits divided into three subclasses like N-methyl d-aspartate (NDMA) receptor, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate receptors.
-
3.
P2X receptor family, which has three subunits of cysteine loop. These are cation permeable-gated channels and opened or closed by ATP interaction (Fig. 6.49).
Fig. 6.49 
Diagrammatic representation of ligand-gated ion channel subclasses on the structural subunit basis. (1) The pentameric cysteine loop superfamily receptor superfamily. (2) The tetrameric ionotropic glutamate receptors are subdivided into N-methyl-d-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate receptor subfamilies. The highly schematic topography of each receptor category indicates the locations of the extracellular and intracellular termini, the number of transmembrane spans (large colored cylinders), and cysteine residues participating in disulfide bond formation (yellow circles). Red cylinders indicate α-helical regions participating in ion conduction/selectivity

6.18 Transport Through Ion Channel P2X Receptors
The trimeric ATP-activated channels are permeable to cations like sodium, potassium, and calcium. There are seven subclasses of the group, which are involved in various physiological functions like muscle contraction, neurotransmitter release, immune responses’ regulation. P2X receptors are present only in eukaryotes and ubiquitously expressed. P2X are trimers of any three from seven variable subunits (P2x1-7) and exist as homomer or heteromer. Each of the subunit may have two transmembranes. The detail is discussed in Chap. 9.
6.19 The Pentamer Cysteine Loop Gama-Amino Butyric Acid Receptors (GABAA)
In nerve transmission, the neurotransmitter is released in synaptic cleft by presynaptic vesicles. These neurotransmitters have to be internalized by postsynaptic membrane for the propagation of impulses. For internalization, these neurotransmitters bind to receptors on postsynaptic membrane and induce conformational change in receptor to open channel for translocation. The GABA and glycine receptors are anion channels but inhibitory in nature. GABA receptors have two subdivisions as GABA receptors family of ligand-gated channel (ionotropic) GABAA and G receptor protein family (GABAB). The G receptors are discussed in Chaps. 3 and 9.
The ligand for GABAA receptor is γ-aminobutyrate. The cysteine loop family in eukaryotes has conserved motif of two cysteine separated by 13 other amino acids. The GABAA receptors are multisubunits with major isoform of GABAA.
Receptors are α1β2 and γ2. The various isoforms of these proteins are found in human as α-subunits with six isoforms, β-subunits three, γ-subunits three, ρ-subunits three, and ∈-, δ-, θ-, and π-subunits one copy. The single subunit of GABAA receptor has around 500 amino acids with both C-terminal and N-terminal that are extracellular. Its half hydrophilic N-terminal has signature motif of cysteine, and other C-terminal has four transmembrane helices from M1 to M4. The M2 transmembrane forms lining of channel. The large intracellular loop between M3 and M4 constitutes modulation site for phosphorylation. The M3 and M4 domains of receptor may interact with other regulatory proteins. The functional receptors are made up of five subunits as homopentamer or heteropentamer. The ϵ- and δ-subunits enhance polymeric association of monomer (Fig. 6.50).

A GABAA receptor major isoform assembly α1β2γ2. The five subunits are arranged in periphery to form central ion pore. a Topology of a single subunit. b Top view of the pentamer. c The various arrangements in a pentamer of subunits three different types, α (yellow), β (red), and γ (green)
The density of these receptors varies from cell to cell. The GABAa are found in nerve cells, muscle, and liver and on immune cells. The GABA receptors initially were thought to be only on postsynaptic membrane for opening of GABA chloride channel for hyperpolarization of a depolarized membrane but recent studies have confirmed their presence on dendrites, and other tissues also for the regulation of synapses. The GABAA receptors are gated chloride anion channels and occasionally may translocate bicarbonate anions. GABA binds to two binding sites at the interface of α-/β-subunit of receptor. The important region for ion translocation is α-subunit segment (F loop), which bulges out from ligand binding site toward the ion channel, and in γ-subunit, this segment modulates channel activity. The second segment, a lysine residue of the short loop between M2 and M3, is important for the formation of a salt bridge during the process of gating. The third segment resides within the β4–β5 linker of the β-subunit. GABA binding to binding sites at the interface of α-/β-subunit stimulates conformational change around binding site which passes through open and many closed states (flipped states) for final gate opening.
6.20 Tetrameric Ionotropic Glutamate Receptor Channels N-Methyl-d-Aspartate (NMDA)
The N-methyl-d-aspartate (NMDA) receptors are known as glutamate-gated cation channel for calcium permeation. The tetrameric ionotropic receptors include NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate receptor as per their synthetic ligand binding. These are excitatory neurotransmitters. In excitatory transmission, glutamate released from presynaptic membrane binds to NDMA or AMPA receptor of postsynaptic membrane. The NDMA receptor family have three different subunits known as Glu N1-3. The crystallographic and functional studies have revealed detail structure and mechanism of channel opening. Active NMDA receptors are tetramer with two GluN1 and two GluN2. The monomer subunit may be divided into four distinct domains as extracellular N-terminal domain, ligand binding domain, pore-forming membrane domain, and C-terminal intracellular. The extracellular N-terminal domain is linked to ligand binding domain through small linker, which is also attached to pore-forming transmembrane (Fig. 6.51).

Various domain representations of NMDAR. a The arrangement of NMDAR subunits with four hydrophobic domains (M1 through M4), two ligand binding domains (S1 and S2), and amino and carboxyl terminal domains (NTD, CTD). b Monomer subunit with N-terminal signal peptide on extracellular side, transmembrane helices M1, M3, and M4, M2, a cytoplasmic reentrant hairpin loop a link between M1 and M3 and ligand binding domains S1 and S2, c Two-dimensional membrane folding model of glycine binding NR1 and glutamate binding NR2 subunits
The transmembrane is liked to C-terminal domain. The channel opening starts in sequential order of ligand binding \(\to\) change in conformation \(\to\) channel opening. Two separate ligands are required for NMDA receptors activation. The ligand binding domain of subunit has two subdomains S1 and S2. The S1 domain of two neighboring subunits, at their interface, creates ligand binding site and is rigid in structure but lower one S2 domain is mobile without any interaction. In typical NDMA receptor, GluN1 and GluN2 will interact with two molecules of glutamate and two molecules of glycine. The receptor with Glu N1 and Glu N3 will require only glycine for the activation. The NMDA receptors have high affinity to glutamate, high calcium (Ca2+) permeability, which may be blocked by magnesium ion in voltage-dependent manner. The NMDA differs from AMPA receptor of the same family in its function. The N-methyl-d-aspartate (NMDA) receptors unlike α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA, activation time 2 ms) are slow in activation (app. 10–50 ms) and deactivation (app. 50–500 ms). AMPA receptor initiates rapid polarization in response to neurotransmitter release during synaptic transmission but NMDA receptors only care for time span of synaptic currents. The glycine is coagonist for NMDA receptor. Two molecules of glycine and two molecules of glutamate bind on separate binding site in receptor for the activation and opening of channel, which results in influx of calcium and sodium with efflux of potassium. The electrical synapses are converted into chemical synapses through NMDA receptor because influx of calcium in postsynaptic membrane activates calcium-dependent signaling pathway for the modulation of synaptic structure, plasticity, and connectivity.
The signaling is discussed in Chap. 9. The independent binding of two ligands, glycine and glutamate, collectively induces change in conformation to open channel. The closing of channel may result in glutamic acid release and desensitization of receptors. It has been observed that the NMDA receptors may be in desensitized form with bound glutamate for long period. The other function of NMDA receptor in learning and memory can be inhibited by magnesium ion in voltage-dependent mode. In close state of receptor, the magnesium is bound to receptor in inhibition mode, which can be released only by sufficient depolarization of the membrane. This kind of interaction empowers NMDA receptor to act as coincidence detector for pre- and postsynaptic changes. The slow kinetics of NMDAR-mediated excitatory postsynaptic current (EPSC) supports temporal summation and decreases dendritic filtering of synaptic input. The receptors provide strength to synapses through long-term potentiation (LTP) and weakness by long-term depression (LTD) for learning and memory. Through calcium influx, the overstimulation of NMDA receptors may cause Alzheimer, Parkinson, Huntington, and amyotrophic lateral sclerosis (ALS). Physiologically, NMDA receptors may be desensitized by unfavorable interaction between glycine and glutamate to cause close conformation of receptor or calcium influx activate calmodulin to remove actin 2 from receptor to keep receptor in desensitized mode. The receptors activity may be decreased by zinc, polyamines, and proton. The phosphorylation of receptor protein enhances open form of channel by removing magnesium and inducing favorable conformation.
6.21 Voltage-Gated Ion Channels
The voltage-gated ion channels open or close in response to change in voltage and the unit of membrane potential. In excitable cells like nerve and muscle, the chemical or electrical signaling is initiated through voltage-dependent sodium or calcium influx. The sodium channel protein in human is composed of large α-subunits (260 Kd with 2000 amino acids) and small β-subunits (30–40 Kd); the small β-subunit has two isoforms β-1 and β-2. The sodium channel β-subunits have N-terminal extracellular domain into immunoglobulin-like fold, a single transmembrane segment, and a short intracellular segment. These units may form heterodimer or heterotrimer. The α-subunits are linked noncovalently to β-1 and through disulfide linkage to β-2. The voltage-gated sodium ion channels are also known as NaV channels. In human, α-subunit has various isoforms like Nav1.1, Nav1.2, Nav1.3, Nav1.4, Nav1.5, Nav1.7, Nav1.8, and Nav1.9. The Nav1.7, Nav1.8, and Nav1.9 are found in peripheral nervous system and Nav1.1, Nav1.2, Nav1.3, and Nav1.6 in central nervous system, and the Nav1.5 resides in heart muscle and Nav1.4 in skeletal muscle. The α-subunit is coded by ten different genes in human but one of these codes for nonchannel α-protein is known as sensing helix. β-subunits have four isoforms as β-1, β-2, β-3, and β-4. The β-1 and β-3 associate with α-subunit noncovalently and β-2 and β-4 through disulfide linkage. Navβ-subunits resemble cell adhesion molecules in structure and may have role in localization. The three-dimensional structure of voltage-gated sodium channel has revealed structural detail, mechanism and catalytic amino acids involved, drug target site, and regulatory site. The bacterial sodium channel (NavAb) is seen to have central pore surrounded by four pore-forming helix segments S5 and S6 segments and the intervening pore loop. The four voltage-sensing elements made up of S1–S4 segments are symmetrically associated with the peripheral pore elements.
The structural analysis of bacterial sodium channel (NavAb) has shown the swapping of functional domain of one subunit to other like all individual voltage-sensing elements are found in close associated with the pore-forming element to neighbor as shown in Fig. 6.52 in dark colors. This type of arrangement and association is also observed in potassium channel (Fig. 6.53).

Structure of voltage-dependent bacterial sodium channel (NavAB), a view from upper side, b lateral view, c detail of interacting segments of channel protein

Subunit primary structures of the voltage-gated sodium channels Y, sites of probable N-linked glycosylation; P in red circles, sites of demonstrated protein phosphorylation by PKA (circles) and PKC (diamonds); green, pore-lining segments; white circles, the outer (EEEE) and inner (DEKA) rings of amino residues that form the ion selectivity filter and the drug binding site; yellow, S4 voltage sensors; h in blue circle, inactivation particle in the inactivation gate loop; blue circles, sites implicated in forming the inactivation gate receptor
The NavAb sodium channel are found to have large outer entrance with narrow selectivity filter, a large central cavity filled with water and layered by the S6 segments and an intracellular activation gate at the crossing of the S6 segments and intracellular surface of the membrane. Sodium and potassium channel has similar type of structure but differs significantly in permeation mechanism. The potassium ion channels discriminate K+ by direct interaction with a series of four ion coordination sites created by the carboxyl group of ion selectivity filter amino acid residues in the zone. Water is excluded and does not interact with selection filter, while the NavAb ion selectivity filter has a high-field-strength site for ions at its extracellular end because of the side chains participation of four highly conserved and key determinants glutamate residues. The sodium ion with two planar water molecules for hydration could fit in this high-field-strength site. On outer side of the field, two carboxylic groups of peptide create coordination site for sodium binding with four planar waters of hydration. These negatively charged group interaction with sodium removes water of hydration partially for permeating sodium as a hydrated ion, which interact on the way with the pore residues through its inner shell of bound waters.
The local anesthetics, antiarrhythmic and antiepileptic drugs may inhibit this channel by binding to inner side of segment 6 (drug receptor site) but binding to this site NavAb channels is solely controlled by the side chain of phe 203, single amino acid residue. The sodium channels open in response to depolarization and within fraction of second are closed also for repetitive firing of action potential in synapse. The short intracellular loop connecting α-subunit III and IV domain acts as inactivation gate in channel inactivation. This acts as an intracellular blocking particle by folding into the channel structure and blocks the pore during inactivation. The NMR study of channel inactivation gate has shown the presence of a rigid α-helix followed by two loops of protein with IFM motif and a neighboring threonine residue on their surface for interaction participate in closing. The inactivation gate bends at conserved pair of glycine residues to fold into the intracellular mouth of the pore. The fast inactivation gate is not found in homotetrameric bacterial sodium channels.
The voltage sensing is executed by the S4 transmembrane segments of sodium channels, which has four to eight repeated motifs of a positively charged amino acid residue (usually arginine), followed by two hydrophobic residues, and shifts the gating charges of sodium channels in the sliding helix in voltage sensing. The gating charges are stabilized in their transmembrane position by ionic interacting pairing with neighboring negatively charged residues, which can exchange their ion partners during outward movement.
6.22 K2P Channels Are Not Leaky Channels
The formerly referred leaky channels, resting, back ground, or constitutive channels are now referred as K2P or voltage insensitive channels. The K2P channels are ubiquitously found in all organisms. The functionally active K2P channel has two subunits, and each subunit is made up of two pore regions (P1 and P2), four transmembrane domains (M1–M4), and two characteristic extracellular helices Cap C1 and C2. The Yeast TOK1, KP2 channel with eight transmembrane helices (M1–M8). The TOK1 (2P/8TM) is exceptionally outward rectifier. These channels are highly regulated and functionally important in nervous system, muscles, blood vessels, endocrine system, and kidney. According to sequence homology, eukaryotic K2P channels are classified into six groups; (1) The TWIK clade is weak inward rectifying channels, (2) TREK clade is mechanosensitive and also regulated by lipids, (3) TASK clade channels are regulated by acid pH, (4) TALK clade is alkaline pH-sensitive, (5) THIK clade is inhibited by halothane, and (6) TRESK clade is present in spinal cord.
Unlike voltage-dependent channels (Nav, Kv, Cav), these K2P channel do not have voltage-sensing domain and have two pores. The K2P are not inactivated during depolarization except TWIK-2. The voltage-dependent channels have one pore made by four transmembranes (Fig. 6.54).

Bacterial voltage-dependent sodium channel selection filter
K2P-channels in the cell membrane can be regulated at the transcriptional level. The presence of Cap helices C1 and C2 is unique to these channels, which vertically extends ~35 Å on the extracellular side of membrane to cover entrance with two lateral portals for K+ ions. The pore-lining helices, M2 and M4, traverse obliquely through the membrane and M1 and M3, and the outer helices are more vertically oriented.
The transmembrane segments of the channel have lateral openings between M3 and M4 facing toward lipid environment. The selectivity filter and the pore helices have fourfold symmetry with inner wide open cavity. The extracellular cap, the central cavity, and the inner opening of channel are found in twofold symmetry unlike other potassium channels. The two subunits were found to be assembled in a simple antiparallel manner. The C1–C2 linkers are linked via a disulfide bridge. The two opposite pore helices and the corresponding pore loops contribute to forming the selectivity filter. The M1-helix of one subunit is adjacent to the end to M2-helix of the other subunit. The C-helix is amphipathic with charged residues facing the cytoplasmic side. The human K2P channel cytosolic end of M4 has two to four charged residues and forms an amphipathic helix.
The cytosolic part of the pore participates in gating and permeation. The crystallized K2P-channel has been found to have lateral opening at the interface between the M2-helix of one subunit and the M4-helix of the other subunit to connect the inner cavity with the hydrophobic core of the cell membrane. The lateral cavity, occupied with the hydrophobic tails of membrane phospholipids, may be important in channel gating because of lipid interaction with M2 and M4 domains and alkyl chains of membrane lipids into the cavity through the lateral opening influences the conductance and the pharmacological properties. Like all voltage-dependent channels, the glycine residues are conserved M2 and the M4 helices of all K2P-channels for creating kink in M2 and the M4 domain around the hinge of G268 for moving M4-helix from a “down” to an “up” configuration for transport, where lateral fenestration is closed and ions are passed through the selectivity filter. In down conformation, the central cavity M4-helix crosses the membrane in such a way that lipids of the central cavity blocks the permeation. K2P-channels are lined up with hydrophobic residues in layer. The opening of lower activation gate promotes the opening of upper inactivation gate, and C-type inactivation is promoted unlike voltage-dependent gate. During depolarization, all K2P-channels have shown immediate current component and following time-dependent component of outward current. This instantaneous component supports all K2P channels to have open probability at negative potentials, and the time-dependent current changes support the regulation of K2P-channels by voltage changes. The steady-state current–voltage relation under physiological conditions (with 4–5 mM K+ extracellular concentration) of K2P channel, studied at depolarized potentials, is much larger than the current flowing through K2P channel at resting membrane potential to support role of K2P channel in action potential configuration. The anesthetics enhance the leakage of ions in neural tissue and decrease excitability. In various tissues, K2P channels are present to perform various functions and may also play role in apoptosis subjected to further analysis (Figs. 6.55 and 6.56).

a The K2P TRAAK channel from human, b diagrammatic representation of two pores and four transmembranes in single unit of K2P channel

Structure of TRAAK K2P channels space-filling model. The two subunits are color coded. The hydrophobic residues of the lateral cavity (in the down state) are highlighted in green
References
Cipriano DJ, Wang Y, Bond S et al (2008) Structure and regulation of the vacuolar ATPases. Biochem Biophys Acta 1777:599–604
Cosentino K, Ros U, Garcia Saez AJ (2015) Assembling the puzzle: oligomerization of alpha pore forming protens in membrane. Biochim Biophy Acta 1858(3):457–466
Dawson Roger JP, Locher KP (2006) Structure of a bacterial multidrug ABC transporter. Nature 443:180–185
Deutscher J, Ake FMD, Derkaoui M et al (2014) The bacterial phosphoenolpyruvate: carbohydrate phosphotransferase system: regulation by protein phosphorylation and phosphorylation-dependent protein-protein interactions. Microbiol Mol Biol Rev 78(2):231–256
Gomes D, Agasse A, Thiebaud P et al (2009) Aquaporins are multifunctional water and solute transporters highly divergent in living organisms. Biochimica Biophysica Acta (BBA) Biomembr 1788(6):1213–1228
Gutmann Daniel AP, Ward A, Urbatsch IL et al (2010) Understanding poly specificity of multidrug ABC transporters: closing in on the gaps in ABCB1. Trends Biochem Sci 35:36–42
Hollenstein K, Frei DC, Locher KP (2007) Structure of an ABC transporter in complex with its binding protein. Nature 446:213–216
Hosaka T, Yoshizawa S, Nakajima Y et al (2008) Structural mechanism for light-driven transport by a new type of chloride ion pump, Non labens marinus rhodopsin-3. Br J Pharmacol 154(8):1680–1690
Kelkar DA, Chattopadhay A (2007) The gramicidin ion channel: a model membrane protein. Biochimica Biophysica Acta (BBA) Biomembr 1768(9):2011–2025
Mathie A, Al Moubarak E, Veale EL (2010) Gating of two pore domain potassium channels. J Physiol 588(17):3149–3156
Morth JP, Pedersen BP, Buch-Pedersen MJ et al (2011) A structural overview of the plasma membrane Na+, K+-ATPase and H+-ATPase ion pumps. Nat Rev Mol Cell Biol 12:60–70
Naftalin RJ, Nicholas Green Y, Cunninghamz P (2007) Lactose permease H1-lactose symporter: mechanical switch or brownian ratchet? Biophys J 92:3474–3491
Palmgren MG, Nissen P (2011) P-type ATPases. Ann Rev Biophys 40:243–266
Postma PW, Lengeler JW, Jacobson GR (2000) Phosphoenolpyruvate: carbohydrate phosphotransferase systems of bacteria. Microbiol Rev 57(3):543–594
Rees DC, Johnson E, Lewinson O (2009) ABC transporters: the power to change. Nat Rev Mol Cell Biol 10:218–227. https://doi.org/10.1038/nrm2646
Renigunta V, Schlichthörl G, Daut J, Arch P (2015) Much more than a leak: structure and function of K2P-channels. Eur J Physiol 467:867–894
Tilley SJ, Saibil HR (2006) The mechanism of pore formation by bacterial toxins. Curr Opin Struct Biol 16:230–236
Verkman AS, Anderson MO, Papadopoulos MC (2014) Aquaporins: important but elusive drug targets. Nat Rev Drug Discov 13:259–277
Vyklicky V, Korinek M, Smejkalova T et al (2014) Structure, function, and pharmacology of NMDA receptor channels. Physiol Res 63(Suppl 1):S191–S203
Wright EM, Loo DD, Hirayama BA (2011) Biology of human sodium glucose transporters. Physiol Rev 91(2):733–794
Weeber EJ, Levy M, Sampson MJ et al (2002) The role of mitochondrial porins and the permeability transition pore in learning and synaptic plasticity. J Biol Chem 277:18891–18897
Wilkens S (2015) Structure and mechanism of ABC transporters. F1000 Prime Rep 7:14. https://doi.org/10.12703/P7-14
Zeth K (2010) Structure and evolution of mitochondrial outer membrane proteins of β-barrel topology. Biochimica Biophysica Acta (BBA) Bioenerg 1797(6–7):1292–1299
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter

{kind=link}
Cite this chapter
Wardhan, R., Mudgal, P. (2017). Membrane Transport. In: Textbook of Membrane Biology. Springer, Singapore. https://doi.org/10.1007/978-981-10-7101-0_6
Download citation
DOI: https://doi.org/10.1007/978-981-10-7101-0_6
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-7100-3
Online ISBN: 978-981-10-7101-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)