
Abstract
MNase-seq allows the genome-wide examination of the nucleosome landscape by determination of nucleosome positioning and occupancy. Typically, native or formaldehyde fixed chromatin is subjected to digestion by micrococcal nuclease (MNase), which degrades linker DNA and yields mainly mono-nucleosomes. The resulting material can be processed directly or can be subjected to an optional chromatin immunoprecipitation step (MNase-ChIP-seq). De-crosslinked and purified DNA is then subjected to next-generation sequencing. The protocol presented here has been tailored for the analysis of nucleosome landscape in the malaria parasite, Plasmodium falciparum, but most steps are directly applicable to other cell types. We also discuss general considerations for experimental design and computational analysis, which are crucial for accurate investigation of the nucleosome landscape.
We’re sorry, something doesn't seem to be working properly.
Please try refreshing the page. If that doesn't work, please contact support so we can address the problem.
We’re sorry, something doesn't seem to be working properly.
Please try refreshing the page. If that doesn't work, please contact support so we can address the problem.
Key words
- Micrococcal nuclease (MNase)
- MNase-seq
- MNase-ChIP-seq
- Nucleosome landscape
- Nucleosome positioning
- Nucleosome occupancy
- AT-rich DNA
- Plasmodium falciparum
1 Introduction
In all eukaryotes, the genetic material is packaged into chromatin. The basic unit of the chromatin is the nucleosome, consisting of two copies of each histone type (H2A, H2B, H3, and H4). However, these nucleosomes do not evenly inhabit the genome, which has profound effects on the chromatin structure and accessibility of the underlying DNA sequences. Transcriptionally silent, heterochromatic regions are constantly occupied by densely packed nucleosomes, while active promoter regions are more accessible and characterized by the combination of a nucleosome-depleted region and a downstream array of well-positioned nucleosomes. The basic layout of the nucleosome landscape is dictated by the sequence preference of the histone octamer (that defines the position of certain nucleosomes) and the interaction between histone octamers (that governs nucleosome spacing). Chromatin-modifying enzymes actively adapt this landscape, which makes the chromatin a highly dynamic structure. The resulting nucleosome landscape is typical to each cell and highly informative in deciphering chromatin function. For comprehensive reviews on nucleosome landscape, see [1, 2].
Micrococcal nuclease (MNase) has been isolated from Staphylococcus aureus in the 1960s and was among the first enzymes used to study the chromatin structure [3]. MNase is an endo/exonuclease, which preferentially digests single-stranded DNA, but can nick double-stranded DNA and digest RNA [4]. Importantly, it has a marked preference for linker DNA over DNA wrapped around the histone octamer or occupied by other proteins, making it an ideal enzyme to isolate nucleosomal DNA. MNase was first used to separate hetero- and eu-chromatin or study chromatin structure of individual (trans)genes, but with the rise of the genomic technologies it quickly became the workhorse to study the nucleosome landscape. MNase-seq utilizes next-generation sequencing technology to obtain millions of short sequence reads from the ends of nucleosomal DNA fragments, which can be used to decipher the position and occupancy of nucleosomes on a genome-wide scale [5, 6].
While several other methods have been developed to study the nucleosome landscape (MPE-seq [7]; ChIP-exo [8]; “hydroxy-radical-seq” [9]; “CAD-seq” [10]), MNase-seq remains to be the most used approach for this purpose. Here, we shortly discuss important considerations for the experimental design, caution for potential artifacts and describe a detailed protocol, which we used to decipher the nucleosome landscape of the malaria parasite, Plasmodium falciparum [11].
1.1 Experimental Considerations
While the MNase-seq procedure is reasonably straightforward, there are some key steps, which can markedly influence the outcome and interpretation of the results. Here, we highlight some points of consideration. A more detailed description of potential artifacts introduced in each of these steps can be found in the supplementary results and discussion of [11].
1.1.1 Formaldehyde Crosslinking and Chromatin Immunoprecipitation
In most MNase-seq protocols formaldehyde crosslinking is used to “fix” protein–DNA interactions and avoid movement of nucleosomes during MNase digestion . While in general this is advisable it is worthwhile to keep in mind that formaldehyde crosslinking preferentially crosslinks GC-rich sequences [12] and can crosslink non-histone proteins to the DNA as well. To specifically select for nucleosomal DNA fragments chromatin immunoprecipitation using an antibody against H3 or H4 is being used (MNase-ChIP-seq). This approach, on the other hand, can artificially alter the recovery of DNA from nucleosomes where the epitope is modified (e.g., carry posttranslational modification) or inaccessible (e.g., masked by other proteins) or even further lower the presence of AT-rich DNA fragments which are washed away due to their lower crosslinking efficiency (see above).
1.1.2 Titration of Digestion Conditions
Perhaps the most delicate part of the MNase-seq protocol is to define the digestion condition that yields mainly mononucleosomal fragments with a small amount of dinucleosomal DNA present. This has to be titrated for nearly all samples individually by varying the time of digestion or enzyme concentration applied. Sequencing of multiple digests from the same chromatin sample, as an alternative, can provide a more robust measure of chromatin accessibility [13]. Importantly, differences in digestion conditions for the individual samples can confound inter-sample comparison of the MNase-seq data. While this can be improved to some extent by standardized sample preparation [14], it is advisable to check and match the “extent” of digestion between samples. In addition, it is recommended to mix small amounts of nuclei from an unrelated species (e.g., yeast ) to each sample in a similar proportion that can be used later on to correct for differences in digestion condition (spike -in control). This is particularly relevant if global differences in nucleosome occupancy are expected between the samples (e.g., upon deletion of a histone chaperone).
1.1.3 Size Selection
In early protocols, mononucleosomal-sized fragments have been selected during the preparation of sequencing libraries. This, however, can introduce unwanted technical variation by cutting out slightly different size ranges from the gel or when comparing samples with slightly different “extents” of digestion and leads to the removal of informative sub-nucleosomal or di-nucleosomal fragments. Instead, we advise omitting the size-selection step and use of paired-end sequencing to enable in silico size selection if necessary (see also later).
1.1.4 MNase Digestion Bias
Arguably, the biggest drawback of using MNase to decipher the nucleosome landscape is the intrinsic preference of this enzyme toward AT-rich sequences [15]. This can lead for example to over-digestion and artificial depletion of AT-rich nucleosomal DNA [11] and hence overestimation of the repulsive effect of AT-rich sequences for nucleosome formation [9]. Different approaches have been proposed to deal with these biases using computational methods [16] or using different reagents to fragment the chromatin [7, 9, 10]. The protocol we present here makes use of a combined MNase and exonuclease III treatment to alleviate some of the biases associated with the use of MNase alone [11, 17].
1.1.5 Biases in Next-Generation Sequencing
Biases during library preparation for next-generation sequencing and mapping of sequence reads can also negatively impact proper interpretation of the MNase-seq data [18]. In particular, the PCR -step common to most library preparation methods can bias against AT- and GC-rich sequences and against longer (e.g., di- or tri-nucleosomal) DNA fragments. Therefore, it is advisable to use PCR-free library preparation methods if possible [19, 20] or minimize the number of PCR cycles, increase the elongation time and use polymerases with reduced bias [11]. In addition, sequencing of fragmented genomic DNA (e.g., sonicated, MNase-digested, or both) can be used to correct for most biases related to library preparation and reduced mappability of sequencing reads [11] and is therefore recommended as a control in any MNase-seq procedures.
PCR can also lead to stochastic over-amplification of individual DNA fragments, which could for example lead to artificial “highly positioned” nucleosomes. While in silico removal of duplicate reads (i.e., sequence reads that start and end at the same position) is a commonly used approach to deal with this problem, in case of MNase-seq it can potentially lower the signal from very highly positioned nucleosomes. Therefore, the use of unique molecular identifiers (UMIs, [21]) to uniquely label each DNA fragment with a random sequence tag prior to PCR amplification is the most appropriate approach to deal with this problem.
1.2 Data Analysis
The computational analysis of MNase-seq data deserves a discussion that is beyond the volume of this chapter. Therefore, here we only list some typical steps in data analysis and like to refer our readers to excellent reviews on the subject [22, 23]. Sequence reads obtained from MNase-seq libraries are first mapped against the reference genome to generate a coverage plot [22]. As mentioned above, we recommend the use of paired-end sequencing, which provides more exact information about the coupling between digestion events and hence the fragment length. As a consequence, paired-end sequencing provides more accurate information on the nucleosome landscape and also enables in silico selection of fragments of various sizes (fragments smaller than 80 bp for example can be used to identify transcription factor binding or chromatin remodeling events [24] while dinucleosomal fragment sizes can be informative to confer linker length). Notably, appropriate use of the gDNA-seq control will require in silico correction of the fragment-length distribution of this control library to match the MNase-seq libraries as in [11]. The local distribution of MNase-seq fragment end- or mid-points can then be used to identify positioned nucleosomes. Comparison between MNase-seq datasets obtained from for example different stages of development can help to pinpoint dynamic changes in the nucleosome landscape [11]. Heat-maps or “averaged” nucleosome profiles can be used to investigate the nucleosome behavior surrounding specific genomic elements (e.g., transcription start sites , transcription factor binding sites, splice sites , or transcription end sites) for example in relation to gene expression. Nonetheless, often tailored analysis will need to be devised to answer specific research questions.
2 Materials
2.1 General Reagents
-
1.
Nuclease-free water.
-
2.
100% glycerol.
-
3.
QIAquick Spin Columns and reagents (Qiagen), or equivalent.
-
4.
Qubit dsDNA HS kit (Invitrogen), or equivalent.
2.2 Formaldehyde Cross-Linking and Nuclei Isolation from P. falciparum Asexual Blood-Stage Parasites
-
1.
P. falciparum asexual RBC stage culture in 5% hematocrit (see Note 1 ).
-
2.
Plasmodipur filter (EuroProxima) (optional: 70% ethanol to sterilize the filter) (see Notes 2 and 3 ).
-
3.
10, 20, or 50 ml Syringe.
-
4.
37% formaldehyde (HCHO) solution (see Note 4 ).
-
5.
1.25 M Glycine solution .
-
6.
Phosphate Buffered Saline (PBS) (chilled to 4 °C).
-
7.
10% saponin solution (see Note 5 ).
-
8.
Cell Lysis Buffer (CLB): 10 mM Tris–HCl pH 8.0, 3 mM MgCl2, 0.2% IGEPAL CA-630/Nonidet P-40, protease inhibitor cocktail (Complete, EDTA-free protease inhibitor cocktail, Roche, see Note 6 ). Chill to 4 °C before use.
-
9.
Cell Lysis Buffer Sucrose (CLBS): CLB with 0.25 M sucrose. Chill to 4 °C before use.
2.3 Chromatin Digestion
-
1.
MNase Storage Buffer: 20 mM HEPES pH 7.6, 50 mM NaCl, 50% glycerol.
-
2.
Micrococcal nuclease (MNase; Worthington Biochemical Cooperation): reconstitute lyophilized powder to 20 units/μl stock in MNase Storage Buffer (see Note 7 , store at −80 °C). Dilute to 0.5 units/μl in MNase Storage Buffer and aliquot to single-use aliquots, store at −80 °C.
-
3.
Exonuclease III (100 units/μl; New England Biolabs).
-
4.
Digestion Buffer (DB): 50 mM Tris–HCl pH 7.4, 4 mM MgCl2, 1 mM CaCl2, 0.075% IGEPAL CA-630/Nonidet P-40, 1 mM DTT (see Note 8 ), protease inhibitor cocktail (Complete, EDTA-free protease inhibitor cocktail, Roche, see Note 6 ). Chill to 4 °C before use.
-
5.
Quenching Solution (QS): 2% Triton X-100, 0.6% SDS, 300 mM NaCl, 6 mM EDTA. Chill to 4 °C before use.
-
6.
Sonication Buffer (SB): 25 mM Tris pH 7.4, 1% Triton X-100, 0.3% SDS, 150 mM NaCl, 3 mM EDTA, 2 mM MgCl2, 0.5 mM CaCl2, protease inhibitor cocktail (Complete, EDTA-free protease inhibitor cocktail, Roche, see Note 6 ). Chill to 4 °C before use.
-
7.
10% sodium dodecyl sulfate (SDS) solution (see Note 9 ).
-
8.
1 M sodium bicarbonate (NaHCO3) solution.
-
9.
5 M sodium chloride (NaCl) solution.
2.4 Chromatin Immunoprecipitation (ChIP): Optional
-
1.
ProtA Dynabeads (Life Technologies).
-
2.
ProtG Dynabeads (Life Technologies).
-
3.
Magnetic rack for Eppendorf tubes (Life Technologies DynaMag™-2, or equivalent).
-
4.
Rotating wheel, at 4 °C.
-
5.
Rotating wheel, at RT.
-
6.
ChIP-grade α-histone-core antibody (see Note 10 ).
-
7.
Beads Wash Buffer (BWB): 20 mM Tris–HCl pH 8.0, 2 mM EDTA, 1% Triton-X100, 0.1% SDS, 150 mM NaCl, protease inhibitor cocktail (Complete, EDTA-free protease inhibitor cocktail, Roche, see Note 6 ), 0.1% BSA (see Note 11 ). Chill to 4 °C before use.
-
8.
ChIP Dilution Buffer (CDB): 20 mM Tris–HCl pH 8.0, 2 mM EDTA, 1% Triton X-100, 150 mM NaCl, protease inhibitor cocktail (Complete, EDTA-free protease inhibitor cocktail, Roche, see Note 6 ). Chill to 4 °C before use.
-
9.
ChIP Wash Buffer 1 (CWB1): 20 mM Tris–HCl pH 8.0, 2 mM EDTA, 1% Triton X-100, 0.1% SDS, 150 mM NaCl. Chill to 4 °C before use.
-
10.
ChIP Wash Buffer 2 (CWB2): 20 mM Tris pH 8.0, 2 mM EDTA, 1% Triton X-100, 0.1% SDS, 500 mM NaCl. Chill to 4 °C before use.
-
11.
ChIP Wash Buffer 3 (CWB3): 10 mM Tris-HCl pH 8.0, 1 mM EDTA. Chill to 4 °C before use.
-
12.
ChIP Elution Buffer (CEB): 1% SDS, 0.1 M NaHCO3. Use at RT.
-
13.
10% Triton X-100.
2.5 Sequencing Library Preparation
For reagents and equipment used during library preparation , we refer to the list provided in [11, 25].
2.6 Equipment
-
1.
Ice-buckets and ice.
-
2.
Shaking, temperature-controlled incubator, at 37 °C.
-
3.
Temperature-controlled centrifuge, at 4 °C.
-
4.
Temperature-controlled benchtop centrifuge, at 4 °C.
-
5.
Benchtop centrifuge, at RT.
-
6.
Temperature-controlled water bath, at 37 °C.
-
7.
Thermometer.
-
8.
Shaking, temperature-controlled thermoblock, at 45 °C.
-
9.
Qubit fluorimeter (Invitrogen), or equivalent.
-
10.
Liquid nitrogen (for snap-freezing).
-
11.
Sonicator (Bioruptor™ Next gen Sonicator (Diagenode)), or equivalent.
-
12.
Glass homogenizer (7 ml) with type B pestle (Kontes).
-
13.
Agarose gel electrophoresis system and imager, or Experion (BioRad), or Bioanalyzer (Agilent), or equivalent, and reagents.
-
14.
DNase/RNase-free, Polyethylene Terephthalate (PET), 15 ml and/or 50 ml centrifugation tubes (see Note 12 ).
-
15.
DNase/RNase-free Reaction Tubes (Eppendorf).
-
16.
DNase/RNase-free filter tips.
3 Methods
Carry out all procedures on ice, unless stated otherwise. Use clean solutions, workspace, and filter tips to avoid contamination (see Note 13 ).
3.1 Formaldehyde Cross-Linking and Nuclear Isolation from P. falciparum Asexual Blood Stage Cultures (See Notes 14 and 15 )
-
1.
Mount the Plasmodipur filter on a syringe (see Notes 2 , 3 , and 16 ).
-
2.
Take the parasite culture from 37 °C, pass through the Plasmodipur filter and collect in a 15 or 50 ml PET centrifugation tube (see Note 12 ). Immediately add 37% formaldehyde solution at a concentration of 1% formaldehyde. For P. falciparum asexual stages, minimally use the number of parasites specified in Table 1.
Table 1 The appropriate amount of blood-stage, asexual P. falciparum parasites minimally required to obtain two aliquots of about 8 μl nuclei for MNase-seq -
3.
Incubate for 15 min at 37 °C while shaking.
-
4.
Quench the reaction by addition of 1.25 M glycine to reach a final concentration of 0.125 M glycine. Transfer the culture to ice.
-
5.
Spin the fixed cells for 5 min, 600 × g at 4 °C.
-
6.
Remove the supernatant and wash the pellet with 1 culture volume of chilled PBS by gentle resuspension. Spin for 5 min, 600 × g at 4 °C.
-
7.
Remove the supernatant and gently resuspend the pellet in 1 culture volume of chilled PBS.
-
8.
Add 10% saponin to obtain a final concentration of 0.05% to lyse the RBCs. Gently turn the tube upside down 6× to mix the solution and place back on ice.
-
9.
Incubate for 1–5 min on ice until lysis is observed (see Note 17 ).
-
10.
Spin the tube for 10 min, 2400 × g at 4 °C.
-
11.
Remove the supernatant and wash the pellet twice with 1 culture volume chilled PBS. Resuspend the pellet well and spin for 10 min, 2400 × g at 4 °C at each wash step (see Notes 18 and 19 ).
-
12.
After the final wash remove as much supernatant as possible to obtain clean isolated P. falciparum parasites (see Note 20 ).
-
13.
Resuspend the isolated parasites in 1/3 culture volume CLB.
-
14.
Gently homogenize with a glass homogenizer on ice, using 10–15 strokes.
-
15.
Prepare a 15 ml PET centrifugation tube containing 1/3 culture volume CLBS.
-
16.
Collect the nuclei in CLB (from step 14) from the homogenizer and carefully layer them over the CLBS solution (see Note 21 ).
-
17.
Spin in a swing-out centrifuge for 10 min at 2400 × g at 4 °C.
-
18.
Carefully remove all supernatant and resuspend the nuclei in 1–2 ml CLB supplemented with 25% glycerol. Aliquot to at least two equally sized aliquots of approximately 5–8 μl worth of nuclei each (see Note 22 ). Spin aliquots for 10 min at 2400 × g 4 °C, remove the supernatant as much as possible, snap-freeze and store at −80 °C (see Note 23 ).
3.2 Chromatin Digestion
3.2.1 “Test” Digestions (See Notes 24 and 25, and Fig. 1)
-
19.
Heat the water bath to 37 °C and check the temperature with a thermometer.
-
20.
Prepare five Eppendorf tubes containing 25 μl QS for each aliquot of frozen nuclei that will be used for test-digestion. Place them on ice.
-
21.
Determine for each pellet the digestion time points that will be tested (see Note 26 ). From each aliquot five different reaction time points can be tested in the range of 3–15 min.
-
22.
Remove the nuclei from −80 °C storage and thaw on ice.
-
23.
Resuspend nuclei in 1 ml DB each and spin for 16 min at 2400 × g at 4 °C.
-
24.
Remove all supernatant and resuspend the nuclei in 150 μl DB.
-
25.
Add to each 150 μl reaction 0.5 units MNase and 100 units Exonuclease III. Mix by tapping and place in water bath. Start your timer directly.
-
26.
Keep the nuclei in suspension by briefly tapping the tube every 2 min (see Note 27 ) during the incubation.
-
27.
Once the shortest digestion is completed, mix the nuclei by tapping and quickly remove 25 μl solution. Place the remaining digestion reaction back into the 37 °C water bath immediately and add the 25 μl test-digest to one of the Eppendorf tubes containing 25 μl QS and resuspend well. Leave the tube on ice until all digestions are over.
-
28.
Once the next-shortest digestion is completed, again mix the nuclei in DB by tapping and quickly remove 25 μl solution. Place the remaining digestion reaction back into the 37 °C water bath immediately and add the 25 μl test-digest to one of the Eppendorf tubes containing 25 μl QS, resuspend well and put on ice.
-
29.
Repeat step 28 until all five test-digests per aliquot are in QS on ice (see Note 28 ).
-
30.
Fill up the volume in each tube to 300 μl by addition of SB and briefly sonicate the samples to more effectively free the chromatin from the nuclear debris (see Note 29 ).
-
31.
Spin samples for 10 min, 9600 × g, at 4 °C and transfer the supernatant to a new Eppendorf tube (see Note 30 ).
-
32.
To each sample add: 35 μl 10% SDS, 50 μl 1 M NaHCO3, 85 μl 5 M NaCl, 30 μl MQ.
-
33.
De-crosslink overnight at 45 °C, while shaking in a thermoblock (see Note 31 ).
-
34.
Purify the DNA, for example by using QIAquick Spin Columns (Qiagen) (see Note 32 ).
-
35.
Measure the DNA concentration, for example by using dsDNA HS kit (Qubit).
-
36.
Assess digestion rate by analyzing each sample on a 2% agarose gel (use 100-200 ng DNA/sample) or Experion (BioRad) or Bioanalyzer (Agilent) system along a 100 bp DNA ladder.
-
37.
Determine the most optimal digestion condition for each sample as in Fig. 2 (see Note 33 ).
3.2.2 “Real” Digestion (See Notes 34 and 35)
-
38.
Once the most optimal digestion condition for each sample has been determined, the final digestions can be performed.
-
39.
Perform steps 19 and 22–26 identical to what has been done in the test digestions. This time only a single digestion length for the entire 150 μl sample is used.
-
40.
Once the digestion is completed, remove the tube from the 37 °C water bath and immediately add 150 μl QS. Resuspend well and place on ice (see Note 28 ).
-
41.
Sonicate the samples as in step 30.
-
42.
Spin samples for 10 min, 9600 × g, at 4 °C and transfer the supernatant to a new Eppendorf tube (see Notes 30 and 36 ).
-
43.
Take 50 μl of chromatin for de-crosslinking and add: 18.5 μl 10% SDS, 20 μl 1 M NaHCO3, 38.5 μl 5 M NaCl, 73 μl MQ. The remainder of the chromatin could be used for the optional ChIP procedure (see Subheading 3.3 and Note 37 ).
-
44.
Perform steps 33–35.
-
45.
The isolated DNA can be directly used to continue with Subheading 3.4 in case of MNase-seq without ChIP (called “MNase-seq”; for consideration on whether to include ChIP or not, see Subheading 1) (see Note 38 ).
3.3 Chromatin-Immunoprecipitation (ChIP): Optional (See Subheading 1 and Note 39 )
-
46.
Calculate the DNA concentration in each chromatin sample from the DNA concentration measurement of the de-crosslinked chromatin sample from step 44.
-
47.
For each ChIP use 200 ng of chromatin (see Notes 40 and 41 ) and fill up with SB to 50 μl.
-
48.
Add 50 μl CDB to each 50 μl chromatin sample in SB.
-
49.
Add 1 μg antibody to each reaction and rotate at 4 °C overnight.
-
50.
Prepare the beads as follows: Mix 10 μl ProtA beads solution with 10 μl ProtG beads solution for each ChIP reaction (see Note 42 ). Wash the beads twice with 1 ml BWB as follows: add buffer, invert tube 10×, spin 6 s to max 400 × g, place in a magnetic holder for 1 min and remove the liquid without touching the beads. After the last wash reconstitute the beads in CWB1 bringing the total volume back to 20 μl per ChIP reaction.
-
51.
Add 20 μl of well-mixed ProtA/ProtG-bead slurry to each sample and rotate for 2 h at 4 °C.
-
52.
Take samples from rotation wheel, spin 6 s to max 400 × g, place in a magnetic holder for 30 s and remove the supernatant.
-
53.
Wash 1× using CWB1, 2× using CWB2, and 2× using CWB3. For each wash add 400 μl of appropriate wash buffer, rotate for 5 min at 4 °C, spin 6 s to max 400 × g, place in a magnetic holder for 30 s and remove wash buffer.
-
54.
After the final wash, remove all liquid and bring the samples to RT. Then add 200 μl CEB and mix well. Rotate for 20 min at RT.
-
55.
Briefly spin 6 s to max 400 × g at RT, place in a magnetic holder at RT and transfer the eluate to a new Eppendorf tube (keep at RT) (see Note 43 ).
-
56.
To each 200 μl ChIP eluate add: 10 μl 10% SDS, 10 μl 1 M NaHCO3, 60 μl 5 M NaCl, 10 μl 10% Triton X-100, 10 μl H2O (see Note 44 ).
-
57.
To each 100 μl input sample add: 27 μl 10% SDS, 30 μl 1 M NaHCO3, 57 μl 5 M NaCl, 86 μl H2O (see Note 44 ). Alternatively, the input DNA isolated at step 45 can be used here.
-
58.
Decrosslink, purify, and determine the DNA concentration as prescribed in steps 33–35 (see Notes 32 and 45 ).
3.4 Sequencing Library Preparation (See Note 46 )
-
59.
Use 1–10 ng “MNase-seq”, “MNase-ChIP-seq,” and “gDNA control” DNA per library preparation reaction (see Subheading 1 for more details on the controls).
-
60.
For library preparation follow the detailed LADS protocol provided in Hoeijmakers et al. [25]. Include the modifications prescribed in Kensche et al. [11] to allow inclusion of barcoded adapters and KAPA polymerase mediated library amplification and to prevent library size-selection (also see Subheading 1).
4 Notes
-
1.
CAUTION: P. falciparum asexual intra-erythrocytic stages are infectious via blood contact, so special care should be taken when handling sharp objects.
-
2.
Do not use suspensions of higher than 50% red blood cells (RBCs) and do not process more than 12.5 ml packed RBC per Plasmodipur filter to ensure efficient removal of white blood cells.
-
3.
As an alternative for filtering the iRBC culture during the collection for MNase-Seq, human blood can be pre-filtered before addition to the P. falciparum culture. This option is preferred as it will minimize handling time upstream of cross-linking, but can best be performed in combination with a final Percoll gradient centrifugation synchronization step as in [11]. As Plasmodipur filters are not supplied sterile from the manufacturer, the filter can be sterilized by extensive spraying of 70% ethanol solution to the outside of the filter followed by mounting a syringe containing 20 ml 70% ethanol on the filter and passing the ethanol solution through. Subsequently, the filter needs to be washed with 2 × 50 ml sterile MQ and 20 ml medium before filtering the blood solution. All these steps need to be performed in a sterile-flow cabinet.
-
4.
CAUTION: Formaldehyde is toxic by ingestion, skin contact, and inhalation. Therefore, appropriate protection (gloves, lab-coat, safety glasses) and a well-ventilated working environment are required. It is advised to carefully read and follow the detailed safety instructions provided by the manufacturer.
-
5.
CAUTION: Saponin is toxic upon oral or dermal contact of by inhalation. Use of a dusk mask, safety glasses, and gloves is recommended when weighing and dissolving saponin powder.
-
6.
Roche Protease Inhibitor needs to be added fresh.
-
7.
CRITICAL: MNase enzyme solution needs to be aliquoted into single-use aliquots. Avoid freeze–thaw cycles to retain optimal enzyme activity. As enzyme efficiency might vary for each reconstituted batch, make sure to use the same batch enzyme for “test” and “real” digestions .
-
8.
DTT needs to be added fresh.
-
9.
CAUTION: SDS is toxic upon oral or dermal contact of by inhalation. Use of a dusk mask, safety glasses, and gloves is recommended and weighing and dissolving SDS powder needs to be performed in a fume cabinet with appropriate ventilation. For more information, carefully review the manufacturer’s safety instructions.
-
10.
CRITICAL: It is critical to use a ChIP-grade α-histone-core antibody that will precipitate all nucleosomes irrespective of nucleosome variants or post-translational modifications if the aim is to assess the overall nucleosome landscape, for example use α-H3core (Abcam Ab1791, lot GR88948-1) or α-H4core (Abcam Ab17036, lot GR8733-1).
-
11.
BSA should be added fresh.
-
12.
Make sure to use Polyethylene Terephthalate (PET) when collecting P. falciparum-infected red blood cells (RBCs) . When using centrifugation tubes consisting of other types of plastic and/or coating, the (infected)RBCs might adhere to the sides of the tube.
-
13.
CRITICAL: It is critical to work in a clean area, with clean buffers and pipets and while using filter tips to avoid contamination of DNA from other sources in the final sequencing libraries.
-
14.
The protocol for formaldehyde cross -linking and nuclear isolation described here has been optimized for RBCs infected with P. falciparum asexual stages. For any other cell type or life cycle stage, a dedicated protocol should be used.
-
15.
TIMING: Subheading 3.1 will take roughly 3 h to complete. It will take approximately 2 h from the start of the protocol to complete step 12 (isolate P. falciparum parasites) which introduces a potential PAUSE POINT in this procedure.
-
16.
CRITICAL: Removal of contaminating human white blood cells (WBCs) is required to prevent sequencing large amounts of human DNA, which would severely compromise cost-efficiency.
-
17.
Upon lysis of the RBCs , the culture will turn from opaque to a translucent red color. Sometimes this change is difficult to observe. If no shift in transparency is observed continue with centrifugation after 5 min incubation on ice.
-
18.
After saponin lysis and centrifugation, often RBCs are still observed in the pellet (visible as a (partial) bright red pellet). These will generally lyse upon resuspension during the downstream PBS washes. If substantial amounts of RBCs are still part of the pellet after the second wash add a third wash before continuing.
-
19.
After saponin lysis, ring stage parasites often form a whitish layer on top of the pellet. To prevent loss of parasites, take care to not remove too much supernatant on top of the pellet during the wash steps.
-
20.
PAUSE POINT: Parasites could be stored at this stage. To store the isolated parasites, resuspend the pellet in 1 ml PBS supplemented with 25% glycerol, transfer to an Eppendorf tube, spin for 10 min at 2400 × g at 4 °C, remove the supernatant as much as possible, snap-freeze and store at −80 °C for prolonged periods of time. Upon continuation of the protocol, first wash the parasites once in PBS to remove the glycerol and then continue with step 13.
-
21.
Pipet gently and avoid the final “blow-out” from a pipet boy to prevent mixing of the layers. It is important that two layers are formed (CLBS on the bottom, CLB with nuclei on the top) to ensure clean isolation of nuclei and prevent carry-over of contaminants (e.g., hemoglobin).
-
22.
CRITICAL: It is very important to prepare at least 2—but preferably more—aliquots of equal amounts of isolated nuclei. One or more aliquots can be used to test and optimize the digestion time required to generate predominantly mononucleosomal chromatin fragments. The final aliquot(s) should be used for the “real” digestion using the most optimal digestion conditions as determined from the testing-aliquots (see Figs. 1 and 2).
Fig. 1 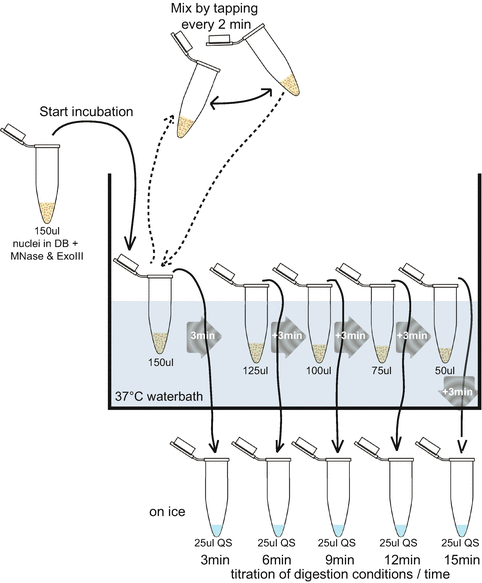
Schematic representation of “test” digestion setup. After addition of MNase and exonuclease III enzymes, the nuclei in digestion buffer (DB) are transferred to a 37 °C water bath. During the 37 °C incubation, regular mixing is required to prevent settling of the nuclei at the bottom of the tube. From a single nuclei aliquot, five different digestion times can be tested by sequentially removing 25 μl from the 150 μl reaction tube at every time point and adding this to 25 μl ice-cold QS
Fig. 2 
2% agarose gel of a typical test digestion of seven tested time-points ranging from 4 to 14 min. Mononucleosomal, dinucleosomal, and trinucleosomal bands are highlighted and the two most optimal digestion times are boxed. Overdigestion is evident from loss-of-sharpness of and increased smearing below the mononucleosomal DNA band
-
23.
PAUSE POINT: Nuclei can be stored at −80 °C for years.
-
24.
CRITICAL: The test digestions are a critical part of the procedure to optimize the right digestion conditions for each batch of nuclei or batch of MNase. This step in the protocol will prevent over- or under-digestion of individual chromatin samples and ensure comparable digestion between samples within a set to allow comparison of those samples. Failure to control (equal) digestion efficiency can result in artifacts as outlined in Subheading 1 (also see Supplementary Results and Discussion in [11]). To ensure equal conditions between “test” and “real” digestions warrant that nuclear aliquots are of equal size, the MNase enzyme is aliquoted from the same batch, the same water bath is used.
-
25.
TIMING: The “Test” digestions in Subheading 3.2 will take roughly 2–3 h “handling” time followed by a 20–24 h overnight incubation and another 2–4 h “handing” time.
-
26.
The enzyme amounts and digestion times indicated in the protocol are examples based on our experience and will likely vary depending on the biological material and the enzyme source. Therefore, each researcher will need to determine this for his-/herself.
-
27.
To prevent nuclei to settle at the bottom of the tube while incubating in the water bath, it is important to regularly mix the tubes by gentle tapping. Make sure to minimize the time that the tubes are out of the 37 °C water bath to allow efficient digestion. Alternatively, a shaking temperature-controlled heat block could be employed. However, in our experience using a heat block less consistent results are obtained, especially for longer digestion times – likely the consequence of increased evaporation to the non-heated lid – therefore the use of a water bath is recommended.
-
28.
PAUSE POINT: Quenched digestion reactions can be stored at −20 °C for prolonged periods of time.
-
29.
The settings of the sonicator will depend on the machine available. We made use of a Bioruptor™ Next gen Sonicator (Diagenode) for six cycles of 10 s ON/10 s OFF using setting LOW (reach level 4). For the Bioruptor sonicator it is advised to transfer the sample to special TPX polymethylpentene tubes (Diagenode 1.5 ml cat# C30010010-300).
-
30.
PAUSE POINT: Soluble, digested chromatin can be stored at 4 °C overnight or at −20 °C for prolonged periods of time.
-
31.
For de-crosslinking procedure, prolonged overnight incubation for 20–24 h is advised to enhance efficiency of de-crosslinking. De-crosslinking at 45 °C instead of 65 °C is advised to reduce sequence bias . For more details see Supplementary Fig. 3 in [25].
-
32.
PAUSE POINT: Purified DNA can be stored at −20 °C for prolonged periods of time.
-
33.
Digestion efficiency of each test digestion can be assessed using 2% agarose gels (always load a 100 bp ladder in parallel). The optimal digestion condition results in primarily mononucleosomes (~150 bp size), with little di- or even trinucleosomal bands present. However, it is important that heavy over-digestion of the mononucleosomal band – characterized by increased smearing to lower size range products – is prevented. For an example test digestion see Fig. 2.
-
34.
It is best practice to handle samples that need to be directly compared on the same day, using the same reagents and conditions.
-
35.
TIMING: The “Real” digestion in Subheading 3.2 will take roughly 2 h “handling” time followed by a 20–24 h overnight incubation and another 30 min “handing” time.
-
36.
CONTROL: To check whether specific chromatin domains remain associated with insoluble nuclear structures (e.g., membranes), the nuclear debris resulting after step 31 may be resuspended in 50 μl SB and de-crosslinked overnight as in step 33 (the pellet can either be used directly for de-crosslinking or can be stored at −20 °C for prolonged periods). The DNA-content from this material can be quantified to estimate the chromatin fraction that remains associated with insoluble nuclear structures and (a subset of) these samples can be further processed into sequencing libraries to assess whether specific chromatin domains become depleted during the procedure (MNase-Pellet-Seq). For P. falciparum, we have found that centromeres are preferentially retained in the insoluble pellet fraction thereby cautioning strong statements on nucleosome occupancy from these regions.
-
37.
PAUSE POINT: Store the remainder of the chromatin as back-up or for later use in MNase-ChIP reactions. Snap-freeze the chromatin and store at −80 °C for years. Make aliquots to avoid many freeze–thaw-cycles.
-
38.
QUALITY CONTROL: Load an aliquot of the DNA on a 2% agarose gel (use 100–200 ng DNA/sample) or Experion (BioRad) or Bioanalyzer (Agilent) system to check digestion efficiency, but ensure that sufficient material is left for sequencing library preparation of “MNase-seq input” samples.
-
39.
TIMING : Subheading 3.3 will take roughly 30 min “handling” time followed by a ~16 h overnight incubation, ~4 h “handing” time, another overnight incubation (20–24 h) and finally ~30 min “handing” time. A PAUSE POINT can be introduced after step 55 (ChIP elution).
-
40.
If the chromatin amount is insufficient to use 200 ng DNA per ChIP reaction less chromatin might be used. To make all reactions comparable also reduce the amount of chromatin added to each reaction from the other chromatin samples.
-
41.
CONTROL: Keep at least 100 ng chromatin aside as input. Fill input sample up to 50 μl by addition of SB and then add 50 μl of CDB, store at 4 °C until de-crosslinking in step 57. Alternatively, use the DNA isolated in step 45 as input sample.
-
42.
CONTROL: For each chromatin sample, besides performing the ChIP reaction, always include a negative control reaction (with negative IgG antibody). The ChIP and control reactions can be assessed by ChIP-qPCR and clear enrichment over negative control should be evident. In our hands, recoveries of negative control samples range 0.1–0.2% recovery for any chromatin region, whereas recoveries for a α-H3core (Abcam Ab1791, lot GR88948-1) or α-H4core (Abcam Ab17036, lot GR8733-1) were in the range of 40% or 3% recovery, respectively.
-
43.
PAUSE POINT: Eluted chromatin can be stored at −20 °C for prolonged periods of time. High concentrations of SDS result in precipitation under low-temperature conditions, therefore ensure to thaw the sample at RT until all precipitated SDS is back in solution before continuing on the protocol.
-
44.
Do not make a pre-mix of the “to-be-added” solutions as the high concentration of salt and detergent will cause precipitation in solution. Instead just add each component separately to each tube.
-
45.
QUALITY CONTROL: Perform a qPCR on the ChIP-material to verify that the ChIP procedure worked properly and confirm that the α-histone ChIP yielded recoveries in the range 3–40% (depending on the α-histone-core antibody used) while the negative IgG control ChIP yielded low recoveries (<0.5%).
-
46.
TIMING: Subheading 3.4 will take approximately 1 day if a KAPA PCR amplification protocol is employed [11] or 2 days if the LADS procedure is employed [25]. Sequencing will take 2–5 days depending on the cycle number, sequencing of single or both ends and the type of sequencer used.


References
Lieleg C, Krietenstein N, Walker M, Korber P (2015) Nucleosome positioning in yeasts: methods, maps, and mechanisms. Chromosoma 124:131–151
Hughes AL, Rando OJ (2014) Mechanisms underlying nucleosome positioning in vivo. Annu Rev Biophys 43:41–63
Clark RJ, Felsenfeld G (1971) Structure of chromatin. Nat New Biol 229:101–106
Heins JN, Suriano JR, Taniuchi H, Anfinsen CB (1967) Characterization of a nuclease produced by Staphylococcus aureus. J Biol Chem 242:1016–1020
Cui K, Zhao K (2012) Genome-wide approaches to determining nucleosome occupancy in metazoans using MNase-Seq. Methods Mol Biol 833:413–419
Platt JL, Kent NA, Harwood AJ, Kimmel AR (2013) Analysis of chromatin organization by deep sequencing technologies. Methods Mol Biol 983:173–183
Ishii H, Kadonaga JT, Ren B (2015) MPE-seq, a new method for the genome-wide analysis of chromatin structure. Proc Natl Acad Sci U S A 112:E3457–E3465
Rhee HS, Bataille AR, Zhang L, Pugh BF (2014) Subnucleosomal structures and nucleosome asymmetry across a genome. Cell 159:1377–1388
Brogaard K, Xi L, Wang JP, Widom J (2012) A map of nucleosome positions in yeast at base-pair resolution. Nature 486:496–501
Allan J, Fraser RM, Owen-Hughes T, Keszenman-Pereyra D (2012) Micrococcal nuclease does not substantially bias nucleosome mapping. J Mol Biol 417:152–164
Kensche PR, Hoeijmakers WA, Toenhake CG, Bras M, Chappell L, Berriman M, Bartfai R (2016) The nucleosome landscape of Plasmodium falciparum reveals chromatin architecture and dynamics of regulatory sequences. Nucleic Acids Res 44:2110–2124
Orlando V, Strutt H, Paro R (1997) Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods 11:205–214
Mieczkowski J, Cook A, Bowman SK, Mueller B, Alver BH, Kundu S, Deaton AM, Urban JA, Larschan E, Park PJ, Kingston RE, Tolstorukov MY (2016) MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat Commun 7:11485
Rizzo JM, Bard JE, Buck MJ (2012) Standardized collection of MNase-seq experiments enables unbiased dataset comparisons. BMC Mol Biol 13:15
Horz W, Altenburger W (1981) Sequence specific cleavage of DNA by micrococcal nuclease. Nucleic Acids Res 9:2643–2658
Kaplan N, Hughes TR, Lieb JD, Widom J, Segal E (2010) Contribution of histone sequence preferences to nucleosome organization: proposed definitions and methodology. Genome Biol 11:140
Nikitina T, Wang D, Gomberg M, Grigoryev SA, Zhurkin VB (2013) Combined micrococcal nuclease and exonuclease III digestion reveals precise positions of the nucleosome core/linker junctions: implications for high-resolution nucleosome mapping. J Mol Biol 425:1946–1960
Meyer CA, Liu XS (2014) Identifying and mitigating bias in next-generation sequencing methods for chromatin biology. Nat Rev Genet 15:709–721
Oyola SO, Otto TD, Gu Y, Maslen G, Manske M, Campino S, Turner DJ, Macinnis B, Kwiatkowski DP, Swerdlow HP, Quail MA (2012) Optimizing Illumina next-generation sequencing library preparation for extremely AT-biased genomes. BMC Genomics 13:1
Hoeijmakers WA, Bartfai R, Francoijs KJ, Stunnenberg HG (2010) Linear amplification for deep sequencing. Nat Protoc 6:1026–1036
Kivioja T, Vaharautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, Taipale J (2012) Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods 9:72–74
Quintales L, Vazquez E, Antequera F (2014) Comparative analysis of methods for genome-wide nucleosome cartography. Brief Bioinform 16:576–587
Teif VB (2015) Nucleosome positioning: resources and tools online. Brief Bioinform 17:745–757
Henikoff JG, Belsky JA, Krassovsky K, MacAlpine DM, Henikoff S (2011) Epigenome characterization at single base-pair resolution. Proc Natl Acad Sci U S A 108:18318–18323
Hoeijmakers WA, Bartfai R, Francoijs KJ, Stunnenberg HG (2011) Linear amplification for deep sequencing. Nat Protoc 6:1026–1036
Acknowledgments
The research leading to this protocol has received funding from The Netherlands Organization for Scientific Research (NWO-Vidi 864.11.007 to R.B.) and The National Institutes of Health (EuPathDB-Driving Biological Project subaward # 553539 to R.B.). We would like to acknowledge Dr. Kensche for valuable input and discussions concerning data analysis and Christa Toenhake for proofreading of the manuscript. Furthermore, we would like to thank our colleagues at the Department of Molecular Biology, the Department of Molecular Developmental Biology, and the Department of Medical Microbiology of Radboud University and St. Radboud UMC for support and advice.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Science+Business Media LLC
About this protocol
Cite this protocol
Hoeijmakers, W.A.M., Bártfai, R. (2018). Characterization of the Nucleosome Landscape by Micrococcal Nuclease-Sequencing (MNase-seq). In: Visa, N., Jordán-Pla, A. (eds) Chromatin Immunoprecipitation. Methods in Molecular Biology, vol 1689. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-7380-4_8
Download citation
DOI: https://doi.org/10.1007/978-1-4939-7380-4_8
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-7379-8
Online ISBN: 978-1-4939-7380-4
eBook Packages: Springer Protocols