
Abstract
Hereditary hemochromatosis (HH) is a group of genetic iron overload disorders that manifest with various symptoms, including hepatic dysfunction, diabetes, and cardiomyopathy. Classic HH type 1, which is common in Caucasians, is caused by bi-allelic mutations of HFE. Severe types of HH are caused by either bi-allelic mutations of HFE2 that encodes hemojuvelin (type 2A) or HAMP that encodes hepcidin (type 2B). HH type 3, which is of intermediate severity, is caused by bi-allelic mutations of TFR2 that encodes transferrin receptor 2. Mutations of SLC40A1 that encodes ferroportin, the only cellular iron exporter, causes either HH type 4A (loss-of-function mutations) or HH type 4B (gain-of-function mutations). Studies on these gene products uncovered a part of the mechanisms of the systemic iron regulation; HFE, hemojuvelin, and TFR2 are involved in iron sensing and stimulating hepcidin expression, and hepcidin downregulates the expression of ferroportin of the target cells. Phlebotomy is the standard treatment for HH, and early initiation of the treatment is essential for preventing irreversible organ damage. However, because of the rarity and difficulty in making the genetic diagnosis, a large proportion of patients with non-HFE HH might have been undiagnosed; therefore, awareness of this disorder is important.
Similar content being viewed by others

Introduction
Iron is essential for numerous cellular processes, but in excess it can damage various important organs through the production of reactive oxygen species [1]. In healthy individuals, the amount of iron in the body is strictly controlled by sophisticated systems [2, 3]. If these systems are disrupted, either systemic iron deficiency or iron overload occur [1, 4]. Hereditary hemochromatosis (HH) is the most common genetic iron overload disorder among Caucasians [5]. Since it was first described by Dr. Armand Trousseau in 1865 as diabetes with bronze-colored skin, researchers have been searching the etiologies and pathophysiology of this disorder [6]. Finally, its responsible gene named HFE (chromosomal location: 6p21) was identified by Feder et al. in 1996 [7]. The product of this gene turned out to be a membrane protein belonging to the major histocompatibility complex class I family that hetero-dimerizes with β2-microglobulin. A pathogenic mutation of this gene causing amino acid substitution of C282Y is quite common in Caucasians in the USA and Europe, especially in the Irish population [8]. After the discovery of HFE, researchers discovered several other gene mutations that also cause different types of HH [9,10,11,12]. Unsurprisingly, products of these genes had important roles in maintaining systemic iron homeostasis, and investigations of these genes contributed to our current understanding of the mechanisms of systemic iron homeostasis [13]. This review summarizes the mechanisms of systemic iron homeostasis and the etiology, diagnosis, and treatment of HH.
Iron absorption from the intestine
Throughout the evolution of life, iron has been an essential and precious nutrient for all organisms. Consequently, active mechanisms for excreting iron from the body do not exist in humans, and the amount of iron in the body is controlled at the entrance, i.e., the duodenal enterocytes. In the duodenum, dietary iron is absorbed as at least 3 forms: inorganic non-heme iron, heme-bound iron, and iron incorporated in ferritin [14]. Among them, the main form is inorganic non-heme iron [15]. In the duodenum, the inorganic non-heme Fe3+ is reduced to Fe2+ by the duodenal cytochrome B reductase [16] and imported by the divalent metal transporter 1 (DMT1), which is expressed in the apical membrane of the enterocytes, into the cytoplasm (Fig. 1) [17, 18]. To maintain systemic iron homeostasis, intestinal iron absorption increases in response to iron deficiency and decreases in iron overload. In the enterocytes, cell surface expression of DMT1 is regulated at both transcriptional and post-transcriptional levels. In the promoters of SLC11A2 gene which encodes DMT1, a hypoxia-responsible element (HRE) exists [19,20,21,22]. In the situations of hypoxia or iron deficiency, expression of hypoxia-inducible factors (HIF1α and HIF2α), which interact with the HRE, increases and thereby activates transcription of SLC11A2. After transcription, the stability of the DMT1 messenger RNA is controlled by a mechanism in which interactions of an iron-responsive element (IRE), a stem loop-like structure in the 3′-untraslated region (UTR), and the iron-regulatory proteins (IRP1 and IRP2), the cytoplasmic iron sensors, play crucial roles [23]. In iron deficiency, IRPs bind to the IRE and stabilize the transcript, resulting in upregulation of translation of DMT1; on the other hand, in iron repletion, IRP1 dissociates from the IRE and IRP2 is degraded after ubiquitination, resulting in destabilization and degradation of the DMT1 transcript [24]. Thus, intestinal expression of iron transporter DMT1 is regulated by these transcriptional and post-transcriptional mechanisms upon intracellular iron status.

Iron absorption by enterocytes. In the duodenum, the inorganic non-heme Fe3+ is reduced to Fe2+ by the duodenal cytochrome B reductase (DCYTB) and imported by the divalent metal transporter 1 (DMT1) into the cytoplasm. Then the imported Fe2+ is transported to iron exporter ferroportin (FPN) located in the basolateral membrane of the enterocyte by a chaperone protein poly(rC)-binding protein 2 (PCBP2). After transportation to the portal vein by FPN, Fe2+ is oxidized to Fe3+ by ferroxidases, i.e., hephaestin (HEPH) and ceruloplasmin (CP), and then carried by transferrin (Tf) into the blood stream
Then the imported Fe2+ into the cytosol is transported to the iron exporter ferroportin (FPN) located in the basolateral membrane of the enterocyte by a chaperone protein, poly(rC)-binding protein 2 [25] (Fig. 1). FPN, which is encoded by the SLC40A1 gene, is the only known iron exporter in vertebrate cells [26, 27]. Although the expression of FPN is partially regulated by HRE at the transcriptional level and by the IRP/IRE system at the post-transcriptional level [20, 27, 28], the main regulator of expression of this protein is hepcidin, the central actor in the systemic iron homeostasis described later. After being transported to the portal vein by FPN, Fe2+ is oxidized to Fe3+ by ferroxidases, i.e., hephaestin and ceruloplasmin, and then carried by transferrin in the blood stream (Fig. 1).
Regulation of systemic iron homeostasis by hepcidin
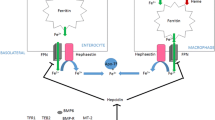
The central regulator of systemic iron homeostasis is hepcidin, a peptide hormone mainly produced by hepatocytes. Hepcidin was discovered by a couple of research groups by means of different approaches [29,30,31,32]. Among them, Ganz’s group originally discovered it as an anti-microbial peptide in the urine [31]. It is transcribed as a proprotein consisting of 84 amino acids, and the peptides of 20–25 amino acids of its carboxyl terminus are cleaved off by furin-like convertases to become mature peptides (Fig. 2). The main form of mature hepcidin consists of 25 amino acids. This active form of hepcidin-25 binds to FPN on the target cells such as enterocytes, macrophages, and hepatocytes, and induces endocytosis and degradation of this iron exporter, thereby reducing iron supply from these cells to the blood stream (Fig. 3) [33]. The expression of hepcidin is regulated by various factors including transferrin-bound iron, iron status of the body, inflammation, hypoxia/anemia, and erythropoietic activity [13]. In iron deficiency, hepcidin expression decreases, which enhances the expression of intestinal expression of FPN, resulting in an increase of iron absorption from the intestine (Fig. 3). In iron-replete conditions, hepcidin expression increases, which downregulates intestinal FPN expression, resulting in a decrease of intestinal iron absorption (Fig. 3). The mechanism by which iron regulates hepcidin expression seems very complex and has not been fully clarified yet, but at least products of HH-responsible genes, HFE, transferrin receptor 2 (TFR2) [9], and hemojuvelin (HJV) [10] are involved in sensing body iron status (Figs. 2, 3).

Regulation of hepcidin expression in the liver. Bone morphogenetic protein 6 (BMP6) secreted by the liver sinusoidal endothelial cells stimulates hepcidin expression through binding to the BMP receptors (BMPRs)/hemojuvelin (HJV) complex and phosphorylates SMAD proteins. The HFE protein/transferrin receptor 2 (TFR2) complex is also involved in iron sensing. The expression of transmembrane protease, serine 6 (TMPRSS6, matriptase 2) increases upon iron deficiency, and it cleaves off the iron-sensing complexes to reduce the expression of hepcidin. Fe–Tf iron-bound transferrin, TFR1 transferrin receptor 1, DMT1 divalent metal transporter 1

Systemic regulation of iron homeostasis. The central regulator of the systemic iron homeostasis is hepcidin which is mainly produced by hepatocytes. There is an iron-sensing complex consisting of bone morphogenetic protein (BMP) receptors (BMPRs), hemojuvelin (HJV), transferrin receptor 2 (TFR2), and HFE protein in the hepatocytes. Upon iron loading, BMP6 and iron-bound transferrin (Fe–Tf) stimulate the promoter of HAMP, the hepcidin gene. Hepcidin downregulates FPN expression, resulting in decreases of intestinal iron absorption and iron release from macrophages
TFR2 is a low-affinity transferrin receptor mainly expressed in the hepatocytes and erythroid precursors [34]. HFE can form a complex with TFR2 in the cell membrane (Figs. 2, 3) [35]. This complex probably plays an important role in sensing transferrin-bound iron. Disruption of either the HFE or TFR2 gene causes downregulation of hepcidin expression [36,37,38]. HJV is a glycosylphosphatidylinositol-anchored membrane protein and a co-receptor for bone morphogenetic proteins (BMPs), which belong to the transforming growth factor-β superfamily [39]. BMP2, 4, 6, and 9 have been shown to induce hepcidin expression [40, 41]. Interaction of BMPs to the BMP receptors causes activation of the promoter of HAMP, the gene encoding hepcidin, through activation of the SMAD phosphorylation pathway (Fig. 2) [41, 42]. In iron-replete mice, hepatic expression of BMP6 increases [43]. In the liver, BMP6 is mainly expressed in the sinusoidal endothelial cells [42, 44] and induces hepcidin expression upon iron loading in a paracrine manner (Fig. 2).
Not only iron, but also other factors regulate hepatic expression of hepcidin. Inflammatory stimuli, e.g., interleukin-6, upregulate hepcidin expression [45]. This regulation is evolutionally developed probably for biological defense, by reducing available iron for the pathogens in the situations of infectious diseases; ironically, this causes iron-restricted anemia observed in chronic inflammation in various clinical settings [46,47,48,49]. In contrast, hypoxia, anemia, and erythropoietic activity downregulate hepcidin expression, thereby increasing iron absorption from the intestine and iron supply from the stored iron in the macrophages [50]. In severe types of thalassemia syndromes and congenital dyserythropoietic anemias, serum levels of hepcidin are inappropriately low (Table 1) and frequently manifest as iron overload even if red blood cell transfusions are not administered [51,52,53]. It is likely that increased erythroblasts in the dyserythropoietic bone marrow secrete a factor that inhibits hepcidin expression. In mice, erythroferrone (FAM132b), a tumor necrosis factor superfamily molecule, was shown to be expressed by erythroblasts and suppress hepcidin expression [54,55,56,57,58].
Various types of HH
The classic type of HH (type 1), caused by mutations of HFE, is common in Caucasians. Common mutations of HFE observed in HH type 1 are H63D, C282Y, and S65C; among them, C282Y is clearly pathogenic [59, 60]. Typically, hepatic dysfunction and diabetes develop in middle-aged individuals, but the disease penetrance is low and only a small proportion of homozygote HFE C282Y mutation manifests symptomatic HH [61]. Men are more susceptible to symptomatic disease than women; the disease penetrance of homozygotes is estimated to be 24–43% in men and 1–14% in women [62]. Genetic background other than HFE and lifestyle habits, such as alcoholic consumption and dietary behavior, can affect susceptibility to HH. Mutation of this gene in humans and disruption of this gene in mice cause iron overload through the reduction of hepcidin expression [37]. HFE protein can interact with TFR1 and TFR2 [35, 63], and it is probably involved in iron sensing in the hepatocytes (Fig. 2).
In contrast to the adult-onset classic type of HH, patients with juvenile types of HH which manifested as systemic iron overload in their early life, although very rare, have been reported [64,65,66]. Manifestations of this type of HH are severe and include hypogonadism, diabetes, and cardiomyopathy [67]. In 1999, a locus of a responsible gene was mapped to 1q21, which was distinct from the locus of HFE [68], and finally a responsible gene, HFE2, was identified in 2004 (HH type 2A, Table 1) [10]. The product of this gene was HJV, which was the co-receptor for BMPs as mentioned previously. Disruption of this gene in mice caused decreased expression of hepcidin and severe iron overload [69], and urine hepcidin levels were low or undetectable in patients with this type of HH despite total body iron excess [70]. Moreover, another type of very severe juvenile HH caused by mutations of HAMP which encodes hepcidin was reported (HH type 2B, Table 1) [11].
In 2000, Camaschella et al. described 6 patients who met the diagnostic criteria for HH but were not linked to HFE from 2 families of Sicilian origin, and identified homozygous Y250X mutation in TFR2 in these patients [9]. Since this discovery, a number of cases with HH harboring various TFR2 mutations have been reported, and this condition is classified as HH type 3 [71, 72]. The Y245X mutation of this gene in mice, equivalent to Y250X in humans, causes downregulation of hepcidin expression and iron accumulation in the liver [36]. Disease severity of this type of HH is moderate.
In patients with HH types 1, 2A, 2B, and 3, the serum hepcidin concentration is inappropriately low despite iron overload, and the diseases are inherited in the autosomal recessive pattern. In contrast, germline mutations of SLC40A1, which encodes FPN, the only cellular iron exporter and target of hepcidin, cause different types of iron overload disorders with appropriately elevated serum hepcidin levels in response to iron excess (Table 1) [73]. Phenotypes of mutations of this gene depend on the positions of mutations [74]; loss-of-function mutations cause HH type 4A (so called “ferroportin disease”) with iron accumulation predominantly in reticuloendothelial cells [75, 76], and gain-of-function mutations cause HH type 4B with accumulation of iron in parenchymal cells [77, 78], similar to HH types 1, 2A, 2B, and 3. In HH type 4A and type 4B, the inheritance pattern is autosomal dominant. The heterozygous mutation of SLC40A1 Q248H is frequently observed in African populations and is associated with mild microcytic anemia and the tendency to iron overload [79, 80].
Recently, families of HH harboring mutations in the propeptide of the BMP6 gene have been reported [81, 82]. According to these studies, serum hepcidin levels of patients with heterozygous mutations of this gene were markedly low or inappropriately low for the iron overload. In addition, Kato et al. described patients with systemic iron overload harboring a heterozygous mutation in the IRE in the 5′-UTR of FTH gene that encodes the ferritin-H subunit from a Japanese family [83]. The proband had iron deposition in hepatocytes and Kupffer cells/macrophages in the liver and spleen. According to their report, expression of the ferritin-H subunit was downregulated and that of ferritin-L subunit was upregulated in the liver of an affected family member, and transfection of the FTH gene with the same mutation caused upregulation of the expression of endogenous ferritin-L subunit in COS-1 cells [83]. Heterozygous FTH knockout mice manifested with hyperferritinemia, but unlike the human cases the mice did not show any sign of iron overload [84].
The prevalence of HFE C282Y mutation is extremely low in Asians [85]. In the literature, I could find only one Japanese hemochromatosis case harboring the homozygous HFE C282Y mutation [86]. However, a Japanese family with a homozygous mutation of HFE (c. 691_693del) responsible for the loss of a tyrosine at the position 231 (p. Y231del) of the HFE protein has been reported [87]. The same mutation was found in the Huh-7 hepatoma cell line and was shown to prevent the translocation of HFE to the cell surface [88]. In addition, families with various types of HH were sporadically reported from Asian countries, including Japan, China, and Pakistan [67, 87, 89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104]. In Japan, mutations in HFE2 (type 2A), TFR2 (type 3), and SLC40A1 (type 4) seem relatively common causes of HH [97].
Hereditary disorders that should be differentiated from HH
Hereditary hyperferritinemia cataract syndrome is caused by mutations of the IRE in the 5′-UTR of the FTL gene, which encodes the ferritin-L subunit. This is an autosomal dominant disorder associated with high serum ferritin levels and the development of cataracts in young individuals without the manifestation of systemic iron overload throughout their lifetime. Dozens of cases have been reported since it was first described in 1995 [105]. Unbalanced formation of ferritin-L and its precipitation in lens was shown to be responsible for the cataracts [106].
Aceruloplasminemia is another rare inherited disorder that should be differentiated from HH. Ceruloplasmin is a serum ferroxidase that converts Fe2+ to Fe3+ ion (Fig. 1). Defects of this enzyme causes iron accumulation in the brain, liver, and pancreas, leading to neurological symptoms, retinal degeneration, diabetes, and anemia [107, 108]. Patients with this disease show low serum copper and iron levels, high serum ferritin levels, and undetectable serum ceruloplasmin. According to the report by Kaneko et al., serum hepcidin levels in patients with this disease were relatively low despite the elevated serum ferritin levels [96].
Hereditary diseases with ineffective erythropoiesis such as thalassemia syndromes, congenital dyserythropoietic anemias, congenital sideroblastic anemias, and hereditary xerocytosis/dehydrated hereditary stomatocytosis also manifest as systemic iron overload due to downregulation of hepcidin expression, and they may need to be differentiated from HH in clinical settings (Table 1) [51, 53, 109,110,111].
Manifestations of HH
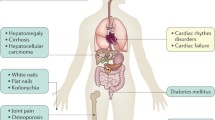
Skin pigmentation, liver dysfunction, diabetes, and cardiomyopathy are common manifestations of HH, which are caused by iron accumulation in the skin, liver, pancreas and heart, respectively (Fig. 4) [112,113,114]. Hepatic dysfunction indicated by elevations of serum alanine aminotransferase levels is often observed in the relatively early stages of this disease. Hepatic dysfunction may progress to cirrhosis and can cause hepatocellular carcinomas [115, 116]. Cardiomyopathy is observed in severe iron overload, and it can cause arrhythmia and heart failure leading to mortality [117]. Iron also accumulates in various endocrine organs and may cause hypothyroidism and hypogonadism [118]. Arthropathy or joint pain is another common but sometimes overlooked symptom of HH [119]. Arthropathy in HH may involve the metacarpophalangeal and proximal interphalangeal joints, wrists, knees, hips, feet, and shoulders, and can be misdiagnosed as rheumatoid arthritis [120, 121]. HH type 4A has distinctive clinical features such as a low transferrin saturation and marginal anemia with low tolerance to phlebotomy [76]. We previously described a Japanese family with HH type 4B and juvenile cataract, though the mechanism of the development of early-onset cataract in this family was undetermined [122].

Symptoms observed in hereditary hemochromatosis
Diagnostic tests
In clinical practice, the most useful biomarker for estimating systemic iron storage is serum ferritin [123]. Ferritin is a cytoplasmic storage protein composed of 24 of ferritin-H and ferritin-L subunits in various ratios, and macrophages actively secrete the ferritin heteropolymer into the blood through a non-classical pathway [124]. If serum ferritin exceeds 300 ng/mL, systemic iron overload is suspected. Caution should be taken to ensure that the expression of ferritin is controlled not only by iron, but also by inflammatory cytokines, hormones, and oxidative stress at both transcriptional and post-transcriptional levels [125]. Therefore, serum ferritin should be evaluated together with C-reactive protein as an inflammatory marker. Remarkable elevation of serum ferritin levels is observed in adult Still’s disease and hemophagocytic syndrome [126, 127].
Transferrin saturation (TSAT), which is calculated by dividing the serum iron level by the total iron binding capacity, is another biomarker for systemic iron status. Iron overload is suspected if TSAT exceeds 50%, and its values over 80% suggests an increase of highly toxic non-transferrin-bound iron (NTBI), which causes organ damage [128]. TSAT does not increase in HH type 4A, so if the serum ferritin level is high and TSAT is low, HH type 4A can be suspected (Table 1).
Imaging tests also help in diagnosing HH. The hepatic density increases on a computed tomography image when excess iron is accumulated. Magnetic resonance imaging (MRI) is another non-invasive way of evaluating iron accumulation in organs. On a T2-intensified MRI scan, the intensity decreases with iron accumulation [129, 130]. For quantification of cardiac iron contents, the T2* method seems more accurate, but this method is not widely available [131]. Liver biopsy is an old and standard method to directly demonstrate iron deposition in the liver, but this procedure is invasive and not routinely performed to make a diagnosis of HH.
In healthy individuals, the serum hepcidin level increases upon iron loading and inflammation and decreases in iron deficiency, hypoxia, and increased erythropoiesis. Therefore, serum levels of hepcidin-25 are positively correlated with the levels of serum ferritin [132, 133]. The serum hepcidin-25 level is abnormally low in types 1, 2A, 2B, and 3 HH for the high serum ferritin levels, and it can be a useful marker for suspecting these types of HH (Table 1). Unfortunately, only selected institutions currently offer this test. A genetic test is necessary for diagnosing HH, and it is widely used for diagnosing HH type 1 [134]. However, examining all the genes known to be responsible for HH requires considerable time and costs. If HH is strongly suspected based on the patient’s clinical features, family history, blood test results, and imaging results, physicians should consult specialists of HH to make a genetic diagnosis [135].
Treatment and management of patients with HH
Early diagnosis and prompt initiation of appropriate treatments are essential for preventing irreversible organ damage [135]. Hepatic function, glucose tolerance, cardiac function, thyroid function, and gonadal function should be evaluated once HH is diagnosed, and symptoms secondary to iron overload, such as diabetes, cardiomyopathy, hypothyroidism, and hypogonadism, should be appropriately treated by consultation with the specialists of these disorders [116]. Phlebotomy is the standard treatment for HH. It should be repeated at appropriate intervals for at least 1 week [116]. Each time before phlebotomy, the hemoglobin (Hb) level should be monitored to avoid inducing anemia. The standard volume of phlebotomy is 400–500 mL, and it should be modified according to the patient’s age, body weight, Hb levels, and comorbidities [116]. The target value of the serum ferritin level is between 50 and 100 ng/mL [136]. Mild liver disease due to iron overload is reversible, but once liver cirrhosis develops it may be irreversible [137]. Improvement of diabetes is often difficult, because once the islet cells are damaged, impaired insulin secretion is rarely recovered [137, 138]. In many patients, insulin therapy is required for treating diabetes. Cardiomyopathy may be reversible by phlebotomy in some patients, but it can be fatal in others [112, 139]. Phlebotomy removes not only iron, but also other factors and nutrients; hence, treatment with erythrocytapheresis in combination with erythropoiesis-stimulating agents experimentally has been reported [140]. In patients with both iron overload and anemia, phlebotomy is inappropriate. In such cases, iron chelation therapy using desferrioxamine, deferasirox, or deferiprone may be considered. Physicians should recommend the patients not to take iron-containing supplements and to avoid alcohol consumption.
Evolutional benefits of HFE mutations
The very high incidences of mutations in the HFE gene among Caucasians evoke the idea that these mutations might have offered advantages to their carriers [114]. For instance, increased iron uptake might have offered advantages to people with a grain-based diet and especially to women of reproductive ages in their survival and reproduction. Unlike C282Y homozygote women who might have hypogonadism, heterozygote women may have a reproducible advantage [141]. Interestingly, a recent study demonstrated that 80% of the French athletes who won international competitions in rowing, Nordic skiing, and judo display mutations in one allele of HFE [142], suggesting that the enhanced iron supply by these mutations might have contributed to their superior physical performance. Mutations of HFE may also modulate the immune system by upregulating expression of FPN and reducing intracellular iron of macrophages, which makes them less susceptible to bacteria and parasites that require iron for their growth [143, 144]. A meta-analysis revealed that a mutation of HFE H63D plays a protective role for Alzheimer disease risk [145]. Large-scale epidemiological studies of Sicilian and Sardinian populations demonstrated that female HFE C282Y heterozygotes had significantly increased life expectancy compared to controls [146, 147]. Taken together, carriers of heterozygous HFE mutations might have some advantages over the wild-type HFE carriers, and these mutations have been inherited in high frequencies.
Conclusions
Hereditary iron overload disorders such as HH and thalassemia syndromes are very common in the world [148,149,150]. Since the discoveries of HFE and hepcidin as the priming pumps in the research field of iron metabolism, a lot of new knowledge about systemic iron regulation have been provided by numerous investigations worldwide. Since the serum ferritin level is not routinely evaluated in clinical practice and because the genetic tests for HH, especially for the non-HFE types, can be performed at only select institutions, a great number of patients with HH with hepatic dysfunction, diabetes, cardiomyopathy, endocrinopathy, and arthropathy might have been undiagnosed and inappropriately treated. If the diagnosis was made in the early stage of the disease, such complications could have been prevented by appropriate treatments, including phlebotomy. Awareness of this disorder is important not only for hematologists, but also for endocrinologists, cardiologists, hepatologists, and especially family physicians.
References
Fleming RE, Ponka P. Iron overload in human disease. N Engl J Med. 2012;366:348–59.
Knutson MD. Iron-sensing proteins that regulate hepcidin and enteric iron absorption. Annu Rev Nutr. 2010;30:149–71.
Gulec S, Anderson GJ, Collins JF. Mechanistic and regulatory aspects of intestinal iron absorption. Am J Physiol Gastrointest Liver Physiol. 2014;307:G397–409.
Miller JL. Iron deficiency anemia: a common and curable disease. Cold Spring Harb Perspect Med. 2013;3:1–13.
Franchini M, Veneri D. Recent advances in hereditary hemochromatosis. Ann Hematol. 2005;84:347–52.
Trousseau A. Glycosurie, diabète sucré. In: Clinique Médicale de l’Hôtel-Dieu de Paris. Vol 2. Paris:J.-B. Baillière et Fils; 1865. p. 663–98.
Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399–408.
Ryan E, O’Keane C, Crowe J. Hemochromatosis in Ireland and HFE. Blood Cells Mol Dis. 1998;24:428–32.
Camaschella C, Roetto A, Cali A, De Gobbi M, Garozzo G, Carella M, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet. 2000;25:14–5.
Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dube MP, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36:77–82.
Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33:21–2.
Wallace DF, Pedersen P, Dixon JL, Stephenson P, Searle JW, Powell LW, et al. Novel mutation in ferroportin1 is associated with autosomal dominant hemochromatosis. Blood. 2002;100:692–4.
Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta. 2012;1823:1434–43.
Masuda T. Soybean ferritin forms an iron-containing oligomer in tofu even after heat treatment. J Agric Food Chem. 2015;63:8890–5.
Anderson GJ, Frazer DM, McKie AT, Vulpe CD, Smith A. Mechanisms of haem and non-haem iron absorption: lessons from inherited disorders of iron metabolism. Biometals. 2005;18:339–48.
McKie AT. The role of Dcytb in iron metabolism: an update. Biochem Soc Trans. 2008;36:1239–41.
Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388:482–8.
Kishi F, Tabuchi M. Complete nucleotide sequence of human NRAMP2 cDNA. Mol Immunol. 1997;34:839–42.
Wang D, Wang LH, Zhao Y, Lu YP, Zhu L. Hypoxia regulates the ferrous iron uptake and reactive oxygen species level via divalent metal transporter 1 (DMT1) Exon1B by hypoxia-inducible factor-1. IUBMB Life. 2010;62:629–36.
Taylor M, Qu A, Anderson ER, Matsubara T, Martin A, Gonzalez FJ, et al. Hypoxia-inducible factor-2alpha mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology. 2011;140:2044–55.
Shah YM, Matsubara T, Ito S, Yim SH, Gonzalez FJ. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab. 2009;9:152–64.
Yeh KY, Yeh M, Polk P, Glass J. Hypoxia-inducible factor-2alpha and iron absorptive gene expression in Belgrade rat intestine. Am J Physiol Gastrointest Liver Physiol. 2011;301:G82–90.
Hubert N, Hentze MW. Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: implications for regulation and cellular function. Proc Natl Acad Sci U S A. 2002;99:12345–50.
Anderson CP, Shen M, Eisenstein RS, Leibold EA. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta. 2012;1823:1468–83.
Yanatori I, Yasui Y, Tabuchi M, Kishi F. Chaperone protein involved in transmembrane transport of iron. Biochem J. 2014;462:25–37.
Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403:776–81.
McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000;5:299–309.
Sangokoya C, Doss JF, Chi JT. Iron-responsive miR-485-3p regulates cellular iron homeostasis by targeting ferroportin. PLoS Genet. 2013;9:e1003408.
Krause A, Neitz S, Magert HJ, Schulz A, Forssmann WG, Schulz-Knappe P, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000;480:147–50.
Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci U S A. 2001;98:8780–5.
Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276:7806–10.
Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–9.
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–3.
Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, Gombart AF, et al. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem. 1999;274:20826–32.
Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem. 2006;281:28494–8.
Kawabata H, Fleming RE, Gui D, Moon SY, Saitoh T, O’Kelly J, et al. Expression of hepcidin is down-regulated in TfR2 mutant mice manifesting a phenotype of hereditary hemochromatosis. Blood. 2005;105:376–81.
Bridle KR, Frazer DM, Wilkins SJ, Dixon JL, Purdie DM, Crawford DH, et al. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet. 2003;361:669–73.
Muckenthaler M, Roy CN, Custodio AO, Minana B, deGraaf J, Montross LK, et al. Regulatory defects in liver and intestine implicate abnormal hepcidin and Cybrd1 expression in mouse hemochromatosis. Nat Genet. 2003;34:102–7.
Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38:531–9.
Truksa J, Peng H, Lee P, Beutler E. Bone morphogenetic proteins 2, 4, and 9 stimulate murine hepcidin 1 expression independently of Hfe, transferrin receptor 2 (Tfr2), and IL-6. Proc Natl Acad Sci U S A. 2006;103:10289–93.
Andriopoulos B Jr, Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet. 2009;41:482–7.
Canali S, Zumbrennen-Bullough KB, Core AB, Wang CY, Nairz M, Bouley R, et al. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood. 2017;129:405–14.
Kautz L, Besson-Fournier C, Meynard D, Latour C, Roth MP, Coppin H. Iron overload induces BMP6 expression in the liver but not in the duodenum. Haematologica. 2011;96:199–203.
Rausa M, Pagani A, Nai A, Campanella A, Gilberti ME, Apostoli P, et al. Bmp6 expression in murine liver non parenchymal cells: a mechanism to control their high iron exporter activity and protect hepatocytes from iron overload? PLoS One. 2015;10:e0122696.
Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101:2461–3.
Kawabata H, Tomosugi N, Kanda J, Tanaka Y, Yoshizaki K, Uchiyama T. Anti-interleukin 6 receptor antibody tocilizumab reduces the level of serum hepcidin in patients with multicentric Castleman’s disease. Haematologica. 2007;92:857–8.
Hohaus S, Massini G, Giachelia M, Vannata B, Bozzoli V, Cuccaro A, et al. Anemia in Hodgkin’s lymphoma: the role of interleukin-6 and hepcidin. J Clin Oncol. 2010;28:2538–43.
Maes K, Nemeth E, Roodman GD, Huston A, Esteve F, Freytes C, et al. In anemia of multiple myeloma, hepcidin is induced by increased bone morphogenetic protein 2. Blood. 2010;116:3635–44.
Song SNJ, Tomosugi N, Kawabata H, Ishikawa T, Nishikawa T, Yoshizaki K. Downregulation of hepcidin resulting from long-term treatment with an anti-IL-6 receptor antibody (tocilizumab) improves anemia of inflammation in multicentric Castleman’s disease (MCD). Blood. 2010;116:3627–34.
Pak M, Lopez MA, Gabayan V, Ganz T, Rivera S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood. 2006;108:3730–5.
Papanikolaou G, Tzilianos M, Christakis JI, Bogdanos D, Tsimirika K, MacFarlane J, et al. Hepcidin in iron overload disorders. Blood. 2005;105:4103–5.
Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13:1096–101.
Kawabata H, Doisaki S, Okamoto A, Uchiyama T, Sakamoto S, Hama A, et al. A case of congenital dyserythropoietic anemia type 1 in a Japanese adult with a CDAN1 gene mutation and an inappropriately low serum hepcidin-25 level. Intern Med. 2012;51:917–20.
Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46:678–84.
Honda H, Kobayashi Y, Onuma S, Shibagaki K, Yuza T, Hirao K, et al. Associations among erythroferrone and biomarkers of erythropoiesis and iron metabolism, and treatment with long-term erythropoiesis-stimulating agents in patients on hemodialysis. PLoS One. 2016;11:e0151601.
Jiang X, Gao M, Chen Y, Liu J, Qi S, Ma J, et al. EPO-dependent induction of erythroferrone drives hepcidin suppression and systematic iron absorption under phenylhydrazine-induced hemolytic anemia. Blood Cells Mol Dis. 2016;58:45–51.
Latour C, Wlodarczyk MF, Jung G, Gineste A, Blanchard N, Ganz T, et al. Erythroferrone contributes to hepcidin repression in a mouse model of malarial anemia. Haematologica. 2017;102:60–8.
Mirciov CS, Wilkins SJ, Dunn LA, Anderson GJ, Frazer DM. Characterization of putative erythroid regulators of hepcidin in mouse models of anemia. PLoS One. 2017;12:e0171054.
Adams PC, Reboussin DM, Barton JC, McLaren CE, Eckfeldt JH, McLaren GD, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 2005;352:1769–78.
Mura C, Raguenes O, Scotet V, Jacolot S, Mercier AY, Ferec C. A 6-year survey of HFE gene test for hemochromatosis diagnosis. Genet Med. 2005;7:68–73.
European Association For The Study Of The L. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol. 2010;53:3–22.
Rossi E, Olynyk JK, Jeffrey GP. Clinical penetrance of C282Y homozygous HFE hemochromatosis. Expert Rev Hematol. 2008;1:205–16.
Lebron JA, Bennett MJ, Vaughn DE, Chirino AJ, Snow PM, Mintier GA, et al. Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell. 1998;93:111–23.
Perkins KW, McInnes IW, Blackburn CR, Beal RW. Idiopathic haemochromatosis in children: report of a family. Am J Med. 1965;39:118–26.
Cazzola M, Ascari E, Barosi G, Claudiani G, Dacco M, Kaltwasser JP, et al. Juvenile idiopathic haemochromatosis: a life-threatening disorder presenting as hypogonadotropic hypogonadism. Hum Genet. 1983;65:149–54.
Camaschella C, Roetto A, Cicilano M, Pasquero P, Bosio S, Gubetta L, et al. Juvenile and adult hemochromatosis are distinct genetic disorders. Eur J Hum Genet. 1997;5:371–5.
Nagayoshi Y, Nakayama M, Suzuki S, Hokamaki J, Shimomura H, Tsujita K, et al. A Q312X mutation in the hemojuvelin gene is associated with cardiomyopathy due to juvenile haemochromatosis. Eur J Heart Fail. 2008;10:1001–6.
Roetto A, Totaro A, Cazzola M, Cicilano M, Bosio S, D’Ascola G, et al. Juvenile hemochromatosis locus maps to chromosome 1q. Am J Hum Genet. 1999;64:1388–93.
Huang FW, Pinkus JL, Pinkus GS, Fleming MD, Andrews NC. A mouse model of juvenile hemochromatosis. J Clin Invest. 2005;115:2187–91.
Nemeth E, Roetto A, Garozzo G, Ganz T, Camaschella C. Hepcidin is decreased in TFR2 hemochromatosis. Blood. 2005;105:1803–6.
Rueda Adel C, Grande NC, Fernandez EA, Enriquez de Salamanca R, Sala LA, Jimenez MJ. Mutations in HFE and TFR2 genes in a Spanish patient with hemochromatosis. Rev Esp Enferm Dig. 2011;103:379–82.
Radio FC, Majore S, Binni F, Valiante M, Ricerca BM, De Bernardo C, et al. TFR2-related hereditary hemochromatosis as a frequent cause of primary iron overload in patients from Central-Southern Italy. Blood Cells Mol Dis. 2014;52:83–7.
De Domenico I, Ward DM, Nemeth E, Vaughn MB, Musci G, Ganz T, et al. The molecular basis of ferroportin-linked hemochromatosis. Proc Natl Acad Sci U S A. 2005;102:8955–60.
Cremonesi L, Forni GL, Soriani N, Lamagna M, Fermo I, Daraio F, et al. Genetic and clinical heterogeneity of ferroportin disease. Br J Haematol. 2005;131:663–70.
Pietrangelo A, Montosi G, Totaro A, Garuti C, Conte D, Cassanelli S, et al. Hereditary hemochromatosis in adults without pathogenic mutations in the hemochromatosis gene. N Engl J Med. 1999;341:725–32.
Pietrangelo A. The ferroportin disease. Blood Cells Mol Dis. 2004;32:131–8.
Njajou OT, Vaessen N, Joosse M, Berghuis B, van Dongen JW, Breuning MH, et al. A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat Genet. 2001;28:213–4.
Montosi G, Donovan A, Totaro A, Garuti C, Pignatti E, Cassanelli S, et al. Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest. 2001;108:619–23.
Gordeuk VR, Caleffi A, Corradini E, Ferrara F, Jones RA, Castro O, et al. Iron overload in Africans and African–Americans and a common mutation in the SCL40A1 (ferroportin 1) gene. Blood Cells Mol Dis. 2003;31:299–304.
Rivers CA, Barton JC, Gordeuk VR, Acton RT, Speechley MR, Snively BM, et al. Association of ferroportin Q248H polymorphism with elevated levels of serum ferritin in African Americans in the Hemochromatosis and Iron Overload Screening (HEIRS) Study. Blood Cells Mol Dis. 2007;38:247–52.
Piubelli C, Castagna A, Marchi G, Rizzi M, Busti F, Badar S, et al. Identification of new BMP6 pro-peptide mutations in patients with iron overload. Am J Hematol. 2017;92:562–8.
Daher R, Kannengiesser C, Houamel D, Lefebvre T, Bardou-Jacquet E, Ducrot N, et al. Heterozygous mutations in BMP6 pro-peptide lead to inappropriate hepcidin synthesis and moderate iron overload in humans. Gastroenterology. 2016;150(672–83):e4.
Kato J, Fujikawa K, Kanda M, Fukuda N, Sasaki K, Takayama T, et al. A mutation, in the iron-responsive element of H ferritin mRNA, causing autosomal dominant iron overload. Am J Hum Genet. 2001;69:191–7.
Ferreira C, Santambrogio P, Martin ME, Andrieu V, Feldmann G, Henin D, et al. H ferritin knockout mice: a model of hyperferritinemia in the absence of iron overload. Blood. 2001;98:525–32.
Distante S, Robson KJ, Graham-Campbell J, Arnaiz-Villena A, Brissot P, Worwood M. The origin and spread of the HFE-C282Y haemochromatosis mutation. Hum Genet. 2004;115:269–79.
Sohda T, Okubo R, Kamimura S, Ohkawara T. Hemochromatosis with HFE gene mutation in a Japanese patient. Am J Gastroenterol. 2001;96:2487–8.
Takano A, Niimi H, Atarashi Y, Sawasaki T, Terasaki T, Nakabayashi T, et al. A novel Y231del mutation of HFE in hereditary haemochromatosis provides in vivo evidence that the Huh-7 is a human haemochromatotic cell line. Liver Int. 2011;31:1593–7.
Vecchi C, Montosi G, Pietrangelo A. Huh-7: a human “hemochromatotic” cell line. Hepatology. 2010;51:654–9.
Imanishi H, Liu W, Cheng J, Ikeda N, Amuro Y, Hada T. Idiopathic hemochromatosis with the mutation of Ala176Val heterozygous for HFE gene. Intern Med. 2001;40:479–83.
Hattori A, Wakusawa S, Hayashi H, Harashima A, Sanae F, Kawanaka M, et al. AVAQ 594-597 deletion of the TfR2 gene in a Japanese family with hemochromatosis. Hepatol Res. 2003;26:154–6.
Kohgo Y. A novel ferroportin disease in a Japanese patient. Intern Med. 2005;44:393–4.
Koyama C, Hayashi H, Wakusawa S, Ueno T, Yano M, Katano Y, et al. Three patients with middle-age-onset hemochromatosis caused by novel mutations in the hemojuvelin gene. J Hepatol. 2005;43:740–2.
Koyama C, Wakusawa S, Hayashi H, Suzuki R, Yano M, Yoshioka K, et al. Two novel mutations, L490R and V561X, of the transferrin receptor 2 gene in Japanese patients with hemochromatosis. Haematologica. 2005;90:302–7.
Koyama C, Wakusawa S, Hayashi H, Ueno T, Suzuki R, Yano M, et al. A Japanese family with ferroportin disease caused by a novel mutation of SLC40A1 gene: hyperferritinemia associated with a relatively low transferrin saturation of iron. Intern Med. 2005;44:990–3.
Hayashi H, Wakusawa S, Motonishi S, Miyamoto K, Okada H, Inagaki Y, et al. Genetic background of primary iron overload syndromes in Japan. Intern Med. 2006;45:1107–11.
Kaneko Y, Miyajima H, Piperno A, Tomosugi N, Hayashi H, Morotomi N, et al. Measurement of serum hepcidin-25 levels as a potential test for diagnosing hemochromatosis and related disorders. J Gastroenterol. 2010;45:1163–71.
Hattori A, Miyajima H, Tomosugi N, Tatsumi Y, Hayashi H, Wakusawa S. Clinicopathological study of Japanese patients with genetic iron overload syndromes. Pathol Int. 2012;62:612–8.
Hattori A, Tomosugi N, Tatsumi Y, Suzuki A, Hayashi K, Katano Y, et al. Identification of a novel mutation in the HAMP gene that causes non-detectable hepcidin molecules in a Japanese male patient with juvenile hemochromatosis. Blood Cells Mol Dis. 2012;48:179–82.
Li S, Xue J, Chen B, Wang Q, Shi M, Xie X, et al. Two middle-age-onset hemochromatosis patients with heterozygous mutations in the hemojuvelin gene in a Chinese family. Int J Hematol. 2014;99:487–92.
Yamashita T, Morotomi N, Sohda T, Hayashi H, Yoshida N, Ochi K, et al. A male patient with ferroportin disease B and a female patient with iron overload similar to ferroportin disease B. Clin J Gastroenterol. 2014;7:260–4.
Ikuta K, Hatayama M, Addo L, Toki Y, Sasaki K, Tatsumi Y, et al. Iron overload patients with unknown etiology from national survey in Japan. Int J Hematol. 2017;105:353–60.
Wang Y, Du Y, Liu G, Guo S, Hou B, Jiang X, et al. Identification of novel mutations in HFE, HFE2, TfR2, and SLC40A1 genes in Chinese patients affected by hereditary hemochromatosis. Int J Hematol. 2017;105:521–5.
Hsiao PJ, Tsai KB, Shin SJ, Wang CL, Lee ST, Lee JF, et al. A novel mutation of transferrin receptor 2 in a Taiwanese woman with type 3 hemochromatosis. J Hepatol. 2007;47(2):303–6.
Lok CY, Merryweather-Clarke AT, Viprakasit V, Chinthammitr Y, Srichairatanakool S, Limwongse C, et al. Iron overload in the Asian community. Blood. 2009;114:20–5.
Girelli D, Corrocher R, Bisceglia L, Olivieri O, De Franceschi L, Zelante L, et al. Molecular basis for the recently described hereditary hyperferritinemia-cataract syndrome: a mutation in the iron-responsive element of ferritin L-subunit gene (the “Verona mutation”). Blood. 1995;86:4050–3.
Brooks DG, Manova-Todorova K, Farmer J, Lobmayr L, Wilson RB, Eagle RC Jr, et al. Ferritin crystal cataracts in hereditary hyperferritinemia cataract syndrome. Invest Ophthalmol Vis Sci. 2002;43:1121–6.
Kono S, Suzuki H, Takahashi K, Takahashi Y, Shirakawa K, Murakawa Y, et al. Hepatic iron overload associated with a decreased serum ceruloplasmin level in a novel clinical type of aceruloplasminemia. Gastroenterology. 2006;131:240–5.
Lozano-Varela M, Carrera-Alonso E, Plaza-Palacios G. Aceruloplasminemia: an entity to consider in patients with anemia. Rev Esp Enferm Dig. 2014;106:360–1.
Tanno T, Noel P, Miller JL. Growth differentiation factor 15 in erythroid health and disease. Curr Opin Hematol. 2010;17:184–90.
Tamary H, Shalev H, Perez-Avraham G, Zoldan M, Levi I, Swinkels DW, et al. Elevated growth differentiation factor 15 expression in patients with congenital dyserythropoietic anemia type I. Blood. 2008;112:5241–4.
Imashuku S, Muramatsu H, Sugihara T, Okuno Y, Wang X, Yoshida K, et al. PIEZO1 gene mutation in a Japanese family with hereditary high phosphatidylcholine hemolytic anemia and hemochromatosis-induced diabetes mellitus. Int J Hematol. 2016;104:125–9.
Murphy CJ, Oudit GY. Iron-overload cardiomyopathy: pathophysiology, diagnosis, and treatment. J Card Fail. 2010;16:888–900.
Utzschneider KM, Kowdley KV. Hereditary hemochromatosis and diabetes mellitus: implications for clinical practice. Nat Rev Endocrinol. 2010;6:26–33.
Hollerer I, Bachmann A, Muckenthaler MU. Pathophysiological consequences and benefits of HFE mutations: 20 years of research. Haematologica. 2017;102(5):809–17.
Kowdley KV. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology. 2004;127:S79–86.
Crownover BK, Covey CJ. Hereditary hemochromatosis. Am Fam Phys. 2013;87:183–90.
Gulati V, Harikrishnan P, Palaniswamy C, Aronow WS, Jain D, Frishman WH. Cardiac involvement in hemochromatosis. Cardiol Rev. 2014;22:56–68.
McNeil LW, McKee LC Jr, Lorber D, Rabin D. The endocrine manifestations of hemochromatosis. Am J Med Sci. 1983;285:7–13.
von Kempis J. Arthropathy in hereditary hemochromatosis. Curr Opin Rheumatol. 2001;13:80–3.
Pascart T, Richette P, Flipo RM. Treatment of nongout joint deposition diseases: an update. Arthritis. 2014;2014:375202.
Husar-Memmer E, Stadlmayr A, Datz C, Zwerina J. HFE-related hemochromatosis: an update for the rheumatologist. Curr Rheumatol Rep. 2014;16:393.
Yamakawa N, Oe K, Yukawa N, Murakami K, Nakashima R, Imura Y, et al. A novel phenotype of a hereditary hemochromatosis type 4 with ferroportin-1 mutation, presenting with juvenile cataracts. Intern Med. 2016;55:2697–701.
Adams PC, Barton JC, Guo H, Alter D, Speechley M. Serum ferritin is a biomarker for liver mortality in the hemochromatosis and iron overload screening study. Ann Hepatol. 2015;14:348–53.
Cohen LA, Gutierrez L, Weiss A, Leichtmann-Bardoogo Y, Dl Zhang, Crooks DR, et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood. 2010;116:1574–84.
Watt RK. The many faces of the octahedral ferritin protein. Biometals. 2011;24:489–500.
ten Kate J, Drenth JP, Kahn MF, van Deursen C. Iron saturation of serum ferritin in patients with adult onset Still’s disease. J Rheumatol. 2001;28:2213–5.
Ravelli A. Macrophage activation syndrome. Curr Opin Rheumatol. 2002;14:548–52.
Sahlstedt L, Ebeling F, von Bonsdorff L, Parkkinen J, Ruutu T. Non-transferrin-bound iron during allogeneic stem cell transplantation. Br J Haematol. 2001;113:836–8.
Kaltwasser JP, Gottschalk R, Schalk KP, Hartl W. Non-invasive quantitation of liver iron-overload by magnetic resonance imaging. Br J Haematol. 1990;74:360–3.
Echeverria JMA, Castiella A, Emparanza JI. Quantification of iron concentration in the liver by MRI. Insights Imaging. 2012;3:173–80.
Anderson LJ, Holden S, Davis B, Prescott E, Charrier CC, Bunce NH, et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J. 2001;22:2171–9.
Tomosugi N, Kawabata H, Wakatabe R, Higuchi M, Yamaya H, Umehara H, et al. Detection of serum hepcidin in renal failure and inflammation by using ProteinChip System. Blood. 2006;108:1381–7.
Sakamoto S, Kawabata H, Kanda J, Uchiyama T, Mizumoto C, Kitano T, et al. High pretransplant hepcidin levels are associated with poor overall survival and delayed platelet engraftment after allogeneic hematopoietic stem cell transplantation. Cancer Med. 2017;6:120–8.
Scotet V, Le Gac G, Merour MC, Mercier AY, Chanu B, Ka C, et al. Impact of HFE genetic testing on clinical presentation of hereditary hemochromatosis: new epidemiological data. BMC Med Genet. 2005;6:24.
Porto G, Brissot P, Swinkels DW, Zoller H, Kamarainen O, Patton S, et al. EMQN best practice guidelines for the molecular genetic diagnosis of hereditary hemochromatosis (HH). Eur J Hum Genet. 2016;24:479–95.
Rombout-Sestrienkova E, van Kraaij MG, Koek GH. How we manage patients with hereditary haemochromatosis. Br J Haematol. 2016;175:759–70.
DeHart MA. Hereditary hemochromatosis: diagnosis and treatment in primary care. Tenn Med. 1999;92:415–7.
Barton JC, Acton RT. Diabetes in HFE hemochromatosis. J Diabetes Res. 2017;2017:9826930.
von Herbay A, Niederau C, Pilichowska M, Reinecke P, Perings C, Vester E, et al. Cardiomyopathy as the cause of death in genetic hemochromatosis. Z Gastroenterol. 1996;34:178–82.
Mariani R, Pelucchi S, Perseghin P, Corengia C, Piperno A. Erythrocytapheresis plus erythropoietin: an alternative therapy for selected patients with hemochromatosis and severe organ damage. Haematologica. 2005;90:717–8.
Bulaj ZJ, Griffen LM, Jorde LB, Edwards CQ, Kushner JP. Clinical and biochemical abnormalities in people heterozygous for hemochromatosis. N Engl J Med. 1996;335:1799–805.
Hermine O, Dine G, Genty V, Marquet LA, Fumagalli G, Tafflet M, et al. Eighty percent of French sport winners in Olympic, World and Europeans competitions have mutations in the hemochromatosis HFE gene. Biochimie. 2015;119:1–5.
Olakanmi O, Schlesinger LS, Britigan BE. Hereditary hemochromatosis results in decreased iron acquisition and growth by Mycobacterium tuberculosis within human macrophages. J Leukoc Biol. 2007;81:195–204.
Paradkar PN, De Domenico I, Durchfort N, Zohn I, Kaplan J, Ward DM. Iron depletion limits intracellular bacterial growth in macrophages. Blood. 2008;112:866–74.
Lin M, Zhao L, Fan J, Lian XG, Ye JX, Wu L, et al. Association between HFE polymorphisms and susceptibility to Alzheimer’s disease: a meta-analysis of 22 studies including 4,365 cases and 8,652 controls. Mol Biol Rep. 2012;39:3089–95.
Lio D, Balistreri CR, Colonna-Romano G, Motta M, Franceschi C, Malaguarnera M, et al. Association between the MHC class I gene HFE polymorphisms and longevity: a study in Sicilian population. Genes Immun. 2002;3:20–4.
Carru C, Pes GM, Deiana L, Baggio G, Franceschi C, Lio D, et al. Association between the HFE mutations and longevity: a study in Sardinian population. Mech Ageing Dev. 2003;124:529–32.
Rund D. Thalassemia 2016: modern medicine battles an ancient disease. Am J Hematol. 2016;91:15–21.
Tantiworawit A, Charoenkwan P, Hantrakool S, Choeyprasert W, Sivasomboon C, Sanguansermsri T. Iron overload in non-transfusion-dependent thalassemia: association with genotype and clinical risk factors. Int J Hematol. 2016;103:643–8.
Krittayaphong R, Viprakasit V, Saiviroonporn P, Siritanaratkul N, Siripornpitak S, Meekaewkunchorn A, et al. Prevalence and predictors of cardiac and liver iron overload in patients with thalassemia: a multicenter study based on real-world data. Blood Cells Mol Dis. 2017;66:24–30.
Acknowledgements
This work was supported in part by a Grant-in-Aids for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (26461400).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Kawabata reports grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan, during the conduct of the study; personal fees from Nippon Shinyaku Co., Ltd., Celgene Corporation, Kyowa Hakko Kirin Co., Ltd., Alexion Pharmaceuticals Inc., Sanofi K.K., Daiichi Sankyo Co., Ltd., Chugai Pharmaceutical Co., Ltd., Novartis Pharma K.K., and Shire Japan K.K., outside the submitted work.
About this article

Cite this article
Kawabata, H. The mechanisms of systemic iron homeostasis and etiology, diagnosis, and treatment of hereditary hemochromatosis. Int J Hematol 107, 31–43 (2018). https://doi.org/10.1007/s12185-017-2365-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2365-3