
Abstract
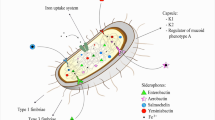
The pathogenesis of Kingella kingae disease begins with colonization of the oropharynx, a process facilitated by type IV pili and a non-pilus trimeric autotransporter adhesin called Knh, factors that mediate adherence to respiratory epithelial cells. A potent RTX cytotoxin with broad cellular specificity may play a role in disrupting the epithelial barrier and facilitating invasion of the bloodstream, possibly in concert with a viral coinfection. Once in the bloodstream, the organism can disseminate to sites of invasive disease, primarily the joints, bones, and endocardium. Survival in the bloodstream and dissemination are likely aided by expression of a capsular polysaccharide and an exopolysaccharide galactan. The evidence for antigenic diversity of K. kingae surface exposed protein epitopes and the observation that type IV pili are selected against during invasive disease suggest that immune system pressure plays an important role in K. kingae pathogenicity.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Colonization
- Adherence
- Pathogenesis
- Type IV pili
- Autotransporter
- Toxin
- Viral coinfection
- Respiratory epithelium
- Capsule
- Exopolysaccharide
- Immunity
- Regulation
Colonization
The pathogenesis of Kingella kingae disease is believed to begin with colonization of the oropharynx. In order to colonize this site, the organism must adhere to respiratory epithelial cells. Early studies demonstrated that K. kingae expresses hair-like surface fibers and suggested that these fibers may be type IV pili, in particular given that they react with antibodies raised against Kingella denitrificans type IV pili. Kehl-fie et al. [1] confirmed that the K. kingae fibers are type IV pili and established that K. kingae type IV pili mediate adherence to respiratory epithelial cells [2]. The density of piliation correlates with three different colony morphologies when K. kingae is grown on solid medium: a spreading/corroding colony type associated with expression of abundant phase variable pili, a non-spreading/noncorroding colony type associated with sparse pili, and a domed colony type associated with no pili [3, 4].
Type IV pili are found in a wide variety of gram-negative species, with examples including enteropathogenic Escherichia coli, Salmonella enterica serovar typhi, Pseudomonas aeruginosa, Legionella pneumophila, Neisseria gonorrhoeae, Neisseria meningitidis, and Vibrio cholerae. These polymeric surface fibers can be up to several μm in length and are composed primarily of a major pilus subunit. Type IV pili major pilus subunits share a number of characteristics, including a short hydrophobic signal peptide that is cleaved during pilus biogenesis, a methylated N-terminal residue, and two cysteine residues near the C-terminus [5]. Structurally, type IV pili major pilus subunits are predicted to have an N-terminal alpha-helical domain and a C-terminal globular domain [6–8].
Type IV pili are unique in their ability to be retracted by the organism [9]. Retraction is responsible for a form of surface motility called twitching motility, allowing the organism to pull itself across a surface as a consequence of sequential rounds of extension, anchoring, and then retraction of pilus fibers [10]. Type IV pilus retraction is also involved in DNA uptake, an essential step in K. kingae natural competence.
Functional expression of type IV can require over 20 gene products, depending on the particular bacterial species. The main steps in type IV pilus biogenesis are as follows: The major pilus subunit (PilA1 in K. kingae) is translated as a pre-protein, which undergoes cleavage of the signal sequence by a dedicated inner membrane peptidase called PilD [11]. After processing, the mature subunit can be incorporated into the growing type IV pilus fiber by the pilus assembly complex, which contains the cytoplasmic protein PilM, the periplasmic proteins PilN, PilO, and PilP, the inner membrane spanning protein PilG, and the oligomeric outer membrane secretin PilQ [12–15]. Pilus extension is powered by ATP hydrolysis by the PilF (PilB in the Pseudomonas type IV pilus nomenclature) cytoplasmic assembly ATPase [16, 17]. Retraction of the pilus through the PilQ secretion is mediated by the PilT retraction ATPase [18, 19].
Gene deletion studies have revealed that pilA1 and pilF are essential for type IV pilus expression in K. kingae [2]. The pilT gene is essential for pilus retraction, and deletion of pilT results in accumulation of dense nonretractile fibers [20]. Representative transmission electron micrographs of negatively stained wild type, pilF mutant, and pilT mutant derivatives of K. kingae strain 269–492 are shown in Fig. 1. Abundant pili are observed emanating from the surface of the wild-type strain (Fig. 1a) and the retraction-deficient pilT mutant (Fig. 1c) but are absent in the pilF assembly mutant (Fig. 1b). K. kingae contains minor pilins called PilA2 and FimB, which are likely incorporated into the pilus fiber but have unclear functions, as elimination of these proteins via deletion of the relevant gene has no effect on type IV pilus phenotypes [2].

Transmission electron micrographs of K. kingae strain 269–492 type IV pili surface fibers. a Abundant pili are seen emanating from the surface of wild-type strain 269–492. b Pili are absent from the surface of an isogenic pilF assembly ATPase mutant. c Mutation of the pilT retraction ATPase results in a hyper-piliated phenotype. Bar, 100 nm
The adhesive components of the K. kingae type IV pilus are likely the PilC1 and PilC2 pilus-associated proteins, which also play a role in pilus biogenesis [2, 21]. Both of these proteins have similarity to the well-characterized PilC1 and PilC2 pilus adhesins of the pathogenic Neisseria sp. and the PilY1 adhesin in P. aeruginosa type IV pili. However, unlike the PilC proteins in the pathogenic Neisseria sp., the K. kingae PilC proteins share limited amino acid sequence homology, with overall identity of 7 % and similarity of 16 % [2]. Elimination of either PilC1 or PilC2 results in a decrease in K. kingae adherence to cultured human epithelial cells, and elimination of both PilC1 and PilC2 results in a non-piliated phenotype [2, 21]. While there is not yet any direct evidence showing that the K. kingae PilC1 and/or PilC2 proteins mediate direct interaction with host cells, recent studies demonstrating that a K. kingae mutant that is able to express pilus fibers lacking PilC1 and PilC2 is non-adherent suggest that these proteins are the adhesive components of the fiber (Porsch and St. Geme, unpublished data). The host cell receptor for K. kingae type IV pili is currently unknown.
Similar to P. aeruginosa PilY1 and the pathogenic Neisseria sp. PilC proteins, both PilC1 and PilC2 in K. kingae contain calcium-biding sites [21–23]. Structural analysis of the PilY1 C-terminal domain showed a β-propeller fold containing a bound calcium ion, and mutational studies of the 9 amino acid calcium-binding pocket revealed that calcium binding is essential for the ability of PilY1 to potentiate PilT-mediated pilus retraction and twitching motility [23]. In K. kingae, both PilC1 and PilC2 contain calcium-binding pockets, with PilC1 containing a 9 amino acid PilY1-like calcium-binding site and PilC2 containing a 12 amino acid calmodulin-like calcium-binding site [21]. Calcium binding is necessary for PilC1 to facilitate twitching motility and adherence but has little to no effect on PilC2 function [21]. Although both PilC1 and PilC2 facilitate twitching motility and adherence, they appear to do so by different mechanisms.
The current model for K. kingae adherence to epithelial cells involves initial adherence by type IV pili, pilus retraction, and subsequent tight adherence mediated by the non-pilus trimeric autotransporter adhesin Knh (K ingella NhhA homolog). Trimeric autotransporters are members of the Type V Secretion family and contain an N-terminal signal sequence, an internal passenger domain, and a C-terminal β-barrel outer membrane anchoring domain [24]. The N-terminal signal sequence directs the protein through the Sec secretion system into the periplasm. The protein then integrates into the outer membrane as a trimer, with the C-terminal outer membrane domains trimerizing to form a pore. The internal passenger domain is translocated through this pore and is displayed on the bacterial surface. Trimerization of the passenger domain is facilitated by a coiled-coil domain adjacent to the outer membrane pore [25].
Knh is a 1783 amino acid protein with a predicted molecular mass of approximately 179 kDa. The N-terminal signal sequence consists of amino acids 1–54, and the C-terminal membrane domain consists of amino acids 1695–1783 [20]. Amino acids 55–1694 constitute the internal passenger domain, which is constructed in a modular manner and contains multiple YadA-like head domains, Trp domains, KG domains, Neck domains, and a single iSneck2 domain, as diagrammed in Fig. 2 using the Domain Annotation of Trimeric Autotransporter Adhesin program (daTAA) [26]. Mutation of knh results in an approximately 50 % reduction in adherence to epithelial cells, and full Knh-mediated adhesive activity is dependent on Knh glycosylation by the cytoplasmic N-linking glycosyltransferase called HMW1CKk (the name HMW1CKk reflects the homology to the well-characterized HMW1C glycosyltransferase in nontypable Haemophilus influenzae) [27]. HMW1CKk modifies a least 32 asparagines throughout the Knh passenger domain [27].

Domain annotation of the trimeric autotransporter adhesin Knh expressed by K. kingae strain 269–492. The structural domain annotation was carried out using the Domain Annotation of Trimeric Autotransporter Adhesin (daTAA) program (toolkit.tuebingen.mpg.de/dataa) [26]
Invasion
The pathogenic model of invasive K. kingae disease postulates that the source of invading bacteria is the colonizing population in the upper respiratory tract. According to this model, it should be possible to isolate genotypically identical bacteria from the oropharynx and the site of invasive disease (i.e., blood, joint fluid, and bone) in patients with invasive K. kingae disease. In support of this model, multiple studies have shown invading organisms to be genetically identical to colonizing isolates in individual patients [28, 29]. For the bacterium to transition from colonizer to invasive pathogen, it must breach the respiratory epithelial barrier in the oropharynx and enter the bloodstream.
Multiple epidemiological studies of patients with invasive K. kingae disease have revealed an association with antecedent or coincident viral infection. Yagupsky et al. [30] found that approximately 50 % of children with invasive K. kingae disease displayed symptoms of upper respiratory tract infection, 15 to 20 % had stomatitis, and 15 to 20 % had diarrhea. In agreement with these findings, a study in France found that 19/21 patients with proven K. kingae osteoarticular infection and only 3/8 patients with non-K. kingae osteoarticular infection had at least one respiratory virus present in the oropharynx, including human rhinovirus (12/21), coronavirus OC43 (4/21), parainfluenza virus 1, 2, 3, or 4 (3/21), enterovirus (2/21), or adenovirus (2/21) [31]. Of note, human rhinovirus infection is known to enhance bacterial adherence to respiratory cells and is associated with invasive bacterial infection [32, 33]. Recently, Houmami et al. [34] described a series of five epidemic and four sporadic K. kingae osteoarticular infections between April and October of 2013 in a French hospital. Of the seven patients in this report who were younger than 24 months of age, five had antecedent hand, foot, and mouth disease and two had antecedent stomatitis. Both patients older than 2 years of age did not have evidence of antecedent or concurrent upper respiratory tract viral infection. Further analysis of an 11-month-old with septic arthritis and antecedent hand, foot, and mouth disease revealed the presence of K. kingae DNA in stored joint fluid and coxsackievirus-A6 in a stored stool specimen [34]. These studies strongly support the hypothesis that upper respiratory tract viral infection contributes to the development of K. kingae invasive disease, possibly related to virus-induced damage to the respiratory epithelium.
In addition to virus-induced damage to the respiratory epithelial barrier, K. kingae may breach the epithelium by means of a potent RTX cytotoxin. The K. kingae RTX toxin is a member of the repeats-in-toxin family and has been shown to be cytotoxic to a wide range of cell types in vitro, including respiratory epithelial cells, synovial cells, macrophage-like cells, and red blood cells [35]. The K. kingae RTX toxin was discovered following the observation that introduction of K. kingae onto a cell monolayer resulted in cell rounding and lysis. A transposon library screen identified a locus with high homology to the Mbx locus in Moraxella bovis that is absent in the less pathogenic Kingella species K. oralis and K. denitrificans [35]. This locus also has high homology to RTX toxins in H. influenzae and N. meningitidis, suggesting that K. kingae acquired the rtx genes through horizontal gene transfer [35]. Testing on a wide range of cultured human cells showed that synovial cells and macrophages are more sensitive to RTX toxin effects than respiratory epithelial cells, although respiratory epithelial cells are still lysed [35]. Additional investigation revealed that the RTX toxin is abundant in K. kingae outer membrane vesicles, which induce cytotoxicity when directly delivered to host cells [36].
The K. kingae RTX toxin contains glycine-rich nonapeptide calcium-binding repeats and is secreted by the type I secretion system [37]. It is a pore forming toxin and forms 0.94 nm radius pores in the host cell membrane that display voltage-dependent gating, with a channel conductance similar to general diffusion [37]. Comparison of a wild type and an isogenic RTX mutant strain in an infant rat model of invasive K. kingae disease revealed decreased virulence by the RTX mutant [38]. The rtx genes are present in all K. kingae strains examined to date, and the rtxA and rtxB genes have proven to be reliable targets for PCR-based detection of K. kingae in clinical samples [39–41].
Immune Evasion
Once K. kingae breaches the respiratory epithelial layer and enters the bloodstream, the organism must be able to evade host immune mechanisms in order to persist and disseminate to sites of invasive disease. While there are only a few studies addressing potential K. kingae immune evasion strategies, current evidence suggests that expression of a polysaccharide capsule and antigenic variation of selected surface-displayed proteins play a role in survival of the bacterium during invasive disease.
Examination of K. kingae strains after 48 h of growth on chocolate agar reveals a striking mucoid colony morphology, consistent with production of a polysaccharide capsule [20]. A number of pediatric pathogens (e.g. N. meningitidis, H. influenzae, and Streptococcus pneumoniae) express a polysaccharide capsule, a layer of surface-associated polysaccharide chains that surround and ‘encapsulate’ the bacterium. Polysaccharide capsules are considered classical virulence factors and have been shown to play many roles in host–pathogen interactions, including masking of antigenic surface epitopes, prevention of complement factor deposition, prevention of phagocytosis, and host mimicry [42]. Disruption of a predicted ctrABCD capsule export operon in K. kingae eliminates the mucoid colony morphology and ablates the polysaccharide capsule, as highlighted in Fig. 3 [20]. Structural analysis of the polysaccharide capsule purified from K. kingae strain 269–492 revealed a repeating unit of N-acetylgalactosamine (GalNAc) and 3-deoxy-D-manno-octulosonic acid (Kdo) with the structure of [→ 3)-β-GalpNAc-(1 → 5)-β-Kdop-(2 →]n [43]. A mutant lacking surface capsule was significantly less virulent in the juvenile rat infection model, demonstrating the likely importance of capsule as a K. kingae virulence factor.

Transmission electron micrographs of thin-sectioned cationic ferritin-stained wild-type K. kingae strain 269–492 (a) and an isogenic ctrA capsule export mutant (b). A layer of electron density due the cationic ferritin interactions with the surface capsular polysaccharide is visible on the surface of the wild-type strain (a) but is absent from the nonencapsulated ctrA mutant (b). Bar, 500 nm
Further study by Starr et al. [44] established that the genetic requirements for encapsulation in K. kingae include the predicted ctrABCD export operon, lipA, lipB, and csaA (capsule synthesis region a gene A). Based on homology to the N. meningitidis lipA and lipB gene products and the Escherichia coli kpsC and kpsS gene products, the K. kingae lipA and lipB gene products likely function as retaining βKdo transferases that create the poly-βKdo linker that connects the polysaccharide to the phosphatidylglycerol membrane anchor [44–46]. Mutational studies revealed that CsaA is a bifunctional glycosyltransferase responsible for synthesizing the GalNAc-Kdo capsule polymer [44].
Examination of a large collection of Israeli healthy carrier and invasive disease isolates revealed a total of four K. kingae capsule types, including the GalNAc-Kdo-containing capsule (designated type a), an N-acetylglucosamine-Kdo-containing capsule with a [→6)-α-GlcpNAc (1→5)-β-Kdop-(2→] repeat unit (designated type b), a ribose-Kdo-containing capsule with a [→3)-β-Ribf-(1→2)-β-Ribf-(1→2)-β-Ribf-(1→4)-β-Kdop-(2→] repeat unit (designated type c), and a GlcNAc-galactose-containing capsule with a [→3)(β-Gal-(1→4)]-β-GlcNAc-(1→3)-α-GlcNAc-1-P-(O→] repeat unit (designated type d) [47] (Starr, Porsch, and St. Geme, submitted). In the Israeli collection of isolates, approximately 96 % of invasive disease isolates expressed either the type a or type b capsule. In contrast, only 70 % of carrier isolates expressed the type a or type b capsule, suggesting that the type a and type b capsule structures may provide a selective advantage for the organism in the bloodstream and contribute to the development of K. kingae invasive disease (Starr, Porsch, and St. Geme, submitted). Given that these initial studies focused on Israeli clinical isolates, further research is necessary to determine the global distribution of K. kingae capsule types.
Porsch et al. [20] demonstrated that the polysaccharide capsule is able to mask the adhesive activity of Knh. When functional pili are present, retraction of these appendages following initial adherence to host cells is thought to displace the polysaccharide capsule, allowing Knh to interact with the host cell and mediate high affinity adherence. A mutant lacking type IV pili but still expressing Knh is non-adherent, presumably because Knh is masked by the polysaccharide capsule. Adherence by this mutant can be restored by eliminating capsule expression, uncovering Knh, and allowing Knh access to host cell receptors [20]. While not demonstrated experimentally, it is likely that the polysaccharide capsule is also able to mask other surface antigens, many of which are smaller than Knh.
In addition to the outer membrane-associated polysaccharide capsule, K. kingae has also been found to secrete a galactofuranose exopolysaccharide homopolymer called PAM galactan. Bendaoud et al. [47] reported a PAM galactan structure of [→3)-β-Galf-(1→6)-β-Galf-(1→]n in K. kingae strain PYKK181, and Starr et al. reported a PAM galactan structure of [→5)-β-Galf-(1→]n in strain 269–492 [43]. The PAM galactan in strain PYKK181 was reported to have broad spectrum biofilm inhibitory activity, which may play a role in dispersal of the organism from a biofilm community or may prevent biofilm formation by competing bacteria in the upper respiratory tract [47]. The potential role of the PAM galactan as a virulence factor has not been investigated, but exopolysaccharides from other gram-negative bacteria have been shown to have protective roles during pathogenesis [48, 49].
K. kingae can cause uncomplicated bacteremia or can disseminate from the bloodstream to joints, bones, or the endocardium [50, 51]. Analysis of a collection of K. kingae systemic isolates showed that strains recovered from the blood of patients with uncomplicated bacteremia were generally piliated but typically expressed relatively few pili [52]. In contrast, strains recovered from joint fluid samples, bone aspirates, or the blood of patients with endocarditis were generally non-piliated [52]. It is possible that low-density piliation facilitates a tropism for joints, bones, and the endocardium and potentiates an inflammatory response, which in turn selects against piliated organisms. Consistent with this possibility, pili promote efficient adherence to cultured synovial cells. Similar to type IV pili expressed by other pathogens such as P. aeruginosa, K. kingae pili are regulated by a transcription factor called σ54 and by the PilS/PilR two-component sensor/regulator system [53]. Mutations in the PilS sensor result in a reduced density of pili, similar to the relative reduction in density of pili observed in isolates recovered from the bloodstream compared to isolates from the posterior pharynx. Mutations in the PilR response regulator completely eliminate piliation, similar to the absence of pili observed in isolates from joints and bones [53]. It is unclear which environmental factors influence the activity of σ54, PilS, and PilR and thereby control the density of piliation in K. kingae. The spreading/corroding colony morphology associated with dense piliation, the non-spreading/non-corroding colony morphology associated with sparse piliation, and the domed colony morphology associated with no pili may reflect the relative activity of PilS and PilR [52].
Immunity
To investigate the role of the host immune system in development of K. kingae disease in healthy individuals, Slonim et al. [54] examined serum IgA and IgG levels against outer membrane proteins in 19 children with invasive disease. As expected, there were significant increases in serum IgA and IgG levels in children convalescing from invasive disease. Further study revealed that the age incidence of disease is inversely correlated with serum IgA and IgG levels in healthy individuals. Infants younger than 6 months of age have undetectable levels of IgA but high levels of serum IgG, suggesting that protection from invasive disease in this age group is due to maternally derived IgG [54]. Children 6 to 18 months of age have the highest incidence of disease and the lowest serum IgG levels [55]. Serum IgG and IgA levels in children 2 years of age and older progressively increase while the incidence of disease progressively decreases, suggesting that K. kingae carriage or exposure in the first 2 years of life may be an immunizing event. However, K. kingae outer membrane protein epitopes have been shown to be polymorphic among diverse strains, raising the possibility of strain specific immune responses that may not prevent recolonization by an antigenically distinct strain [56]. In agreement with these studies, many of the K. kingae surface factors examined to date show strain-to-strain variation, suggesting that immune pressure potentially drives antigenic diversity in this species. The PilA1 subunit of type IV pili is expressed by all piliated strains, yet only 52 % of PilA1 amino acid residues are identical across a collection of clinical isolates [52]. Recent evidence also suggests that there is strain-to-strain sequence diversity in Knh and the PilC proteins (Porsch and St. Geme, unpublished data).
References
Weir S, Marrs CF (1992) Identification of type 4 pili in Kingella denitrificans. Infect Immun 60(8):3437–3441
Kehl-Fie TE, Miller SE, St. Geme JW 3rd (2008) Kingella kingae expresses type IV pili that mediate adherence to respiratory epithelial and synovial cells. J Bacteriol 190(21):7157–7163. doi:10.1128/JB.00884-08
Froholm LO, Bovre K (1972) Fimbriation associated with the spreading-corroding colony type in Moraxella kingii. Acta Pathol Microbiol Scand B Microbiol Immunol 80(5):641–648
Henriksen SD (1969) Corroding bacteria from the respiratory tract. 1. Moraxella kingii. Acta Pathol Microbiol Scand 75(1):85–90
Strom MS, Lory S (1993) Structure-function and biogenesis of the type IV pili. Annu Rev Microbiol 47:565–596. doi:10.1146/annurev.mi.47.100193.003025
Craig L, Pique ME, Tainer JA (2004) Type IV pilus structure and bacterial pathogenicity. Nat Rev Microbiol 2(5):363–378. doi:10.1038/nrmicro885
Forest KT, Dunham SA, Koomey M, Tainer JA (1999) Crystallographic structure reveals phosphorylated pilin from Neisseria: phosphoserine sites modify type IV pilus surface chemistry and fibre morphology. Mol Microbiol 31(3):743–752
Parge HE, Forest KT, Hickey MJ, Christensen DA, Getzoff ED, Tainer JA (1995) Structure of the fibre-forming protein pilin at 2.6 A resolution. Nature 378(6552):32–38. doi:10.1038/378032a0
Biais N, Ladoux B, Higashi D, So M, Sheetz M (2008) Cooperative retraction of bundled type IV pili enables nanonewton force generation. PLoS Biol 6(4):e87. doi:10.1371/journal.pbio.0060087
Mattick JS (2002) Type IV pili and twitching motility. Annu Rev Microbiol 56:289–314. doi:10.1146/annurev.micro.56.012302.160938
Nunn DN, Lory S (1991) Product of the Pseudomonas aeruginosa gene pilD is a prepilin leader peptidase. Proc Natl Acad Sci U S A 88(8):3281–3285
Ayers M, Sampaleanu LM, Tammam S, Koo J, Harvey H, Howell PL, Burrows LL (2009) PilM/N/O/P proteins form an inner membrane complex that affects the stability of the Pseudomonas aeruginosa type IV pilus secretin. J Mol Biol 394(1):128–142. doi:10.1016/j.jmb.2009.09.034
Drake SL, Sandstedt SA, Koomey M (1997) PilP, a pilus biogenesis lipoprotein in Neisseria gonorrhoeae, affects expression of PilQ as a high-molecular-mass multimer. Mol Microbiol 23(4):657–668
Martin PR, Hobbs M, Free PD, Jeske Y, Mattick JS (1993) Characterization of pilQ, a new gene required for the biogenesis of type 4 fimbriae in Pseudomonas aeruginosa. Mol Microbiol 9(4):857–868
Tonjum T, Freitag NE, Namork E, Koomey M (1995) Identification and characterization of pilG, a highly conserved pilus-assembly gene in pathogenic Neisseria. Mol Microbiol 16(3):451–464
Turner LR, Lara JC, Nunn DN, Lory S (1993) Mutations in the consensus ATP-binding sites of XcpR and PilB eliminate extracellular protein secretion and pilus biogenesis in Pseudomonas aeruginosa. J Bacteriol 175(16):4962–4969
Nunn D, Bergman S, Lory S (1990) Products of three accessory genes, pilB, pilC, and pilD, are required for biogenesis of Pseudomonas aeruginosa pili. J Bacteriol 172(6):2911–2919
Wolfgang M, Lauer P, Park HS, Brossay L, Hebert J, Koomey M (1998) PilT mutations lead to simultaneous defects in competence for natural transformation and twitching motility in piliated Neisseria gonorrhoeae. Mol Microbiol 29(1):321–330
Wolfgang M, Park HS, Hayes SF, van Putten JP, Koomey M (1998) Suppression of an absolute defect in type IV pilus biogenesis by loss-of-function mutations in pilT, a twitching motility gene in Neisseria gonorrhoeae. Proc Natl Acad Sci U S A 95(25):14973–14978
Porsch EA, Kehl-Fie TE, St. Geme JW 3rd (2012) Modulation of Kingella kingae adherence to human epithelial cells by type IV Pili, capsule, and a novel trimeric autotransporter. MBio 3(5):e00372–e00412
Porsch EA, Johnson MD, Broadnax AD, Garrett CK, Redinbo MR, St. Geme JW 3rd (2013) Calcium binding properties of the Kingella kingae PilC1 and PilC2 proteins have differential effects on type IV pilus-mediated adherence and twitching motility. J Bacteriol 195(4):886–895. doi:10.1128/JB.02186-12
Johnson MD, Garrett CK, Bond JE, Coggan KA, Wolfgang MC, Redinbo MR (2011) Pseudomonas aeruginosa PilY1 binds integrin in an RGD- and calcium-dependent manner. PLoS ONE 6(12):e29629. doi:10.1371/journal.pone.0029629
Orans J, Johnson MD, Coggan KA, Sperlazza JR, Heiniger RW, Wolfgang MC, Redinbo MR (2010) Crystal structure analysis reveals Pseudomonas PilY1 as an essential calcium-dependent regulator of bacterial surface motility. Proc Natl Acad Sci U S A 107(3):1065–1070. doi:10.1073/pnas.0911616107
Cotter SE, Surana NK, St. Geme JW 3rd (2005) Trimeric autotransporters: a distinct subfamily of autotransporter proteins. Trends Microbiol 13(5):199–205. doi:10.1016/j.tim.2005.03.004
Hoiczyk E, Roggenkamp A, Reichenbecher M, Lupas A, Heesemann J (2000) Structure and sequence analysis of Yersinia YadA and Moraxella UspAs reveal a novel class of adhesins. EMBO J 19(22):5989–5999. doi:10.1093/emboj/19.22.5989
Szczesny P, Lupas A (2008) Domain annotation of trimeric autotransporter adhesins–daTAA. Bioinformatics 24(10):1251–1256. doi:10.1093/bioinformatics/btn118
Rempe KA, Spruce LA, Porsch EA, Seeholzer SH, Norskov-Lauritsen N, St. Geme JW 3rd (2015) Unconventional N-linked glycosylation promotes trimeric autotransporter function in Kingella kingae and Aggregatibacter aphrophilus. MBio 6(4):e01206–e01215. doi:10.1128/mBio.01206-15
Yagupsky P, Porat N, Pinco E (2009) Pharyngeal colonization by Kingella kingae in children with invasive disease. Pediatr Infect Dis J 28(2):155–157. doi:10.1097/INF.0b013e318184dbb8
Basmaci R, Ilharreborde B, Bidet P, Doit C, Lorrot M, Mazda K, Bingen E, Bonacorsi S (2012) Isolation of Kingella kingae in the oropharynx during K. kingae arthritis in children. Clin Microbiol Infect 18(5):E134–E136. doi:10.1111/j.1469-0691.2012.03799.x
Yagupsky P, Peled N, Katz O (2002) Epidemiological features of invasive Kingella kingae infections and respiratory carriage of the organism. J Clin Microbiol 40(11):4180–4184
El Houmami N, Minodier P, Dubourg G, Mirand A, Jouve JL, Basmaci R, Charrel R, Bonacorsi S, Yagupsky P, Raoult D, Fournier PE (2015) Patterns of Kingella kingae disease outbreaks. Pediatr Infect Dis J. doi:10.1097/INF.0000000000001010
Ishizuka S, Yamaya M, Suzuki T, Takahashi H, Ida S, Sasaki T, Inoue D, Sekizawa K, Nishimura H, Sasaki H (2003) Effects of rhinovirus infection on the adherence of Streptococcus pneumoniae to cultured human airway epithelial cells. J Infect Dis 188(12):1928–1939. doi:10.1086/379833
Peltola V, Heikkinen T, Ruuskanen O, Jartti T, Hovi T, Kilpi T, Vainionpaa R (2011) Temporal association between rhinovirus circulation in the community and invasive pneumococcal disease in children. Pediatr Infect Dis J 30(6):456–461. doi:10.1097/INF.0b013e318208ee82
El Houmami N, Mirand A, Dubourg G, Hung D, Minodier P, Jouve JL, Charrel R, Raoult D, Fournier PE (2015) Hand, foot and mouth disease and Kingella kingae infections. Pediatr Infect Dis J 34(5):547–548. doi:10.1097/INF.0000000000000607
Kehl-Fie TE, St. Geme JW 3rd (2007) Identification and characterization of an RTX toxin in the emerging pathogen Kingella kingae. J Bacteriol 189(2):430–436. doi:10.1128/JB.01319-06
Maldonado R, Wei R, Kachlany SC, Kazi M, Balashova NV (2011) Cytotoxic effects of Kingella kingae outer membrane vesicles on human cells. Microb Pathog 51(1–2):22–30. doi:10.1016/j.micpath.2011.03.005
Barcena-Uribarri I, Benz R, Winterhalter M, Zakharian E, Balashova N (2015) Pore forming activity of the potent RTX-toxin produced by pediatric pathogen Kingella kingae: characterization and comparison to other RTX-family members. Biochim Biophys Acta 7:1536–1544. doi:10.1016/j.bbamem.2015.03.036
Chang DW, Nudell YA, Lau J, Zakharian E, Balashova NV (2014) RTX toxin plays a key role in Kingella kingae virulence in an infant rat model. Infect Immun 82(6):2318–2328. doi:10.1128/IAI.01636-14
Haldar M, Butler M, Quinn CD, Stratton CW, Tang YW, Burnham CA (2014) Evaluation of a real-time PCR assay for simultaneous detection of Kingella kingae and Staphylococcus aureus from synovial fluid in suspected septic arthritis. Ann Lab Med 34(4):313–316. doi:10.3343/alm.2014.34.4.313
Cherkaoui A, Ceroni D, Emonet S, Lefevre Y, Schrenzel J (2009) Molecular diagnosis of Kingella kingae osteoarticular infections by specific real-time PCR assay. J Med Microbiol 58(Pt 1):65–68. doi:10.1099/jmm.0.47707-0
Lehours P, Freydiere AM, Richer O, Burucoa C, Boisset S, Lanotte P, Prere MF, Ferroni A, Lafuente C, Vandenesch F, Megraud F, Menard A (2011) The rtxA toxin gene of Kingella kingae: a pertinent target for molecular diagnosis of osteoarticular infections. J Clin Microbiol 49(4):1245–1250. doi:10.1128/JCM.01657-10
Willis LM, Whitfield C (2013) Structure, biosynthesis, and function of bacterial capsular polysaccharides synthesized by ABC transporter-dependent pathways. Carbohydr Res 378:35–44. doi:10.1016/j.carres.2013.05.007
Starr KF, Porsch EA, Heiss C, Black I, Azadi P, St. Geme JW 3rd (2013) Characterization of the Kingella kingae polysaccharide capsule and exopolysaccharide. PLoS ONE 8(9):e75409. doi:10.1371/journal.pone.0075409
Starr KF, Porsch EA, Seed PC, St. Geme JW 3rd (2016) Genetic and molecular basis of Kingella kingae encapsulation. Infect Immun 84(6):1775–1784. doi:10.1128/IAI.00128-16
Willis LM, Stupak J, Richards MR, Lowary TL, Li J, Whitfield C (2013) Conserved glycolipid termini in capsular polysaccharides synthesized by ATP-binding cassette transporter-dependent pathways in Gram-negative pathogens. Proc Natl Acad Sci U S A 110(19):7868–7873. doi:10.1073/pnas.1222317110
Willis LM, Whitfield C (2013) KpsC and KpsS are retaining 3-deoxy-D-manno-oct-2-ulosonic acid (Kdo) transferases involved in synthesis of bacterial capsules. Proc Natl Acad Sci U S A 110(51):20753–20758. doi:10.1073/pnas.1312637110
Bendaoud M, Vinogradov E, Balashova NV, Kadouri DE, Kachlany SC, Kaplan JB (2011) Broad-spectrum biofilm inhibition by Kingella kingae exopolysaccharide. J Bacteriol 193(15):3879–3886. doi:10.1128/JB.00311-11
Miajlovic H, Cooke NM, Moran GP, Rogers TR, Smith SG (2014) Response of extraintestinal pathogenic Escherichia coli to human serum reveals a protective role for Rcs-regulated exopolysaccharide colanic acid. Infect Immun 82(1):298–305. doi:10.1128/IAI.00800-13
Simpson JA, Smith SE, Dean RT (1988) Alginate inhibition of the uptake of Pseudomonas aeruginosa by macrophages. J Gen Microbiol 134(1):29–36. doi:10.1099/00221287-134-1-29
Yagupsky P (2004) Kingella kingae: from medical rarity to an emerging paediatric pathogen. Lancet Infect Dis 4(6):358–367. doi:10.1016/S1473-3099(04)01046-1
Yagupsky P, Porsch E, St. Geme JW 3rd (2011) Kingella kingae: an emerging pathogen in young children. Pediatrics 127(3):557–565. doi:10.1542/peds.2010-1867
Kehl-Fie TE, Porsch EA, Yagupsky P, Grass EA, Obert C, Benjamin DK Jr, St. Geme JW 3rd (2010) Examination of type IV pilus expression and pilus-associated phenotypes in Kingella kingae clinical isolates. Infect Immun 78(4):1692–1699. doi:10.1128/IAI.00908-09
Kehl-Fie TE, Porsch EA, Miller SE, St. Geme JW 3rd (2009) Expression of Kingella kingae type IV pili is regulated by sigma54, PilS, and PilR. J Bacteriol 191(15):4976–4986. doi:10.1128/JB.00123-09
Slonim A, Steiner M, Yagupsky P (2003) Immune response to invasive Kingella kingae infections, age-related incidence of disease, and levels of antibody to outer-membrane proteins. Clin Infect Dis 37(4):521–527. doi:10.1086/376913
Dubnov-Raz G, Ephros M, Garty BZ, Schlesinger Y, Maayan-Metzger A, Hasson J, Kassis I, Schwartz-Harari O, Yagupsky P (2010) Invasive pediatric Kingella kingae Infections: a nationwide collaborative study. Pediatr Infect Dis J 29(7):639–643. doi:10.1097/INF.0b013e3181d57a6c
Yagupsky P, Slonim A (2005) Characterization and immunogenicity of Kingella kingae outer-membrane proteins. FEMS Immunol Med Microbiol 43(1):45–50. doi:10.1016/j.femsim.2004.07.002
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 The Author(s)
About this chapter
Cite this chapter
Porsch, E.A., Rempe, K.A. (2016). Pathogenesis of Kingella kingae Disease. In: St. Geme, III, J. (eds) Advances in Understanding Kingella kingae. SpringerBriefs in Immunology. Springer, Cham. https://doi.org/10.1007/978-3-319-43729-3_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-43729-3_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-43728-6
Online ISBN: 978-3-319-43729-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)