
Abstract
Melanoma is the most lethal type of skin cancer. In this study for the first time we analyze a Greek cohort of primary cutaneous melanoma biopsies, subjected to whole exome sequencing, in order to derive their mutational profile landscape. Moreover, in the context of big data analytical methodologies, we integrated the results of the exome sequencing analysis with transcriptomic data of cutaneous melanoma from GEO, in an attempt to perform a multi-layered analysis and infer a tentative disease network for primary melanoma pathogenesis. The purpose of this research is to incorporate different levels of molecular data, so as to expand our understanding of cutaneous melanoma and the broader molecular network implicated with this type of cancer. Overall, we showed that the results of the integrative analysis offer deeper insight in the underlying mechanisms affected by melanoma and could potentially contribute to the valuable effective epidemiological characterization of this disease.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Melanoma is the most dangerous form of skin cancer [1]. Cutaneous melanoma (or melanoma of the skin), the most common type of melanoma, is a complex multi-factorial disease as both environmental and genetic factors are involved in its manifestation [2]. It is often a fatal neoplasm, derived from melanocytes, that accounts for most skin cancer deaths. In the advanced stages of this cancer, therapeutic intervention usually fails to improve survival despite recent advances in immunotherapy. According to the World Health Organization, 132,000 melanoma skin cancers occur globally each year and the global incidence of melanoma continues to increase, with a main predisposal factor; sun exposure.
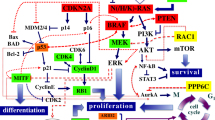
The complexities of cellular metabolism and regulatory pathways involved have, until recently, obstructed the formulation of a unified description for melanoma [3]. Thus, despite the descent of gene signatures for various cancers, e.g. breast or colon cancer, a similar progress remains elusive for malignant melanoma. This could be attributed to the intricate nature of the molecular basis of cutaneous melanoma, which needs neatly stratified epidemiological cohorts to effectively address the issue of the high heterogeneity of this disease. In any case, genomic studies are limited by the shortage of similar melanoma cohorts, collecting and maintaining frozen tumor tissue, therefore rendering gene expression profiling studies of melanoma relatively scarce [4]. Still, efforts have been made to overcome any issues and shed some light on the underlying mechanisms involved with melanoma pathogenesis and metastases [4, 5]. A number of important emerging biological pathways and gene targets recently identified in melanoma are reported in [6]. Key biological pathways, where several significant genes (e.g. CDKN2A, CDK4, RB1) are involved, include proliferation, transcriptional control, extracellular matrix remodeling, glutamate signaling, and apoptosis.
In this study, we have focused on integrating different levels of molecular data through functional analysis to improve our understanding of the underlying mechanisms involved with melanoma. We incorporated established microarray datasets with next generation sequencing mutational data creating a potential disease network for melanoma.
Section 2 describes the techniques and methodology used in this study for the analysis of next generation sequencing and transcriptomic data. Then, in Sect. 3 we present the results derived from the two datasets, in separate subsections, followed by another subsection for data integration. Finally, we conclude this study in Sect. 4.
2 Materials and Methods
2.1 Analysis of Next Generation Exome Sequencing Data
The data analyzed in this section derived from Whole Exome Sequencing (WES) data of paired tumor and adjacent normal tissue from 9 patients with cutaneous melanoma (manuscript under preparation). The framework for the analysis of Next Generation Sequencing (NGS) data includes various state-of-the-art tools and has been previously presented by our team [7].
We first align the reads to the reference genome (hg19, version b37), using BWA (Burrows-Wheeler Aligner) [8] for DNA reads, adjusted for paired-end sequencing and run in consecutive steps for finding the correct coordinates and generate the final alignment in proper format. Then, we preprocess reads using Picard [9], for marking duplicate reads and sorting sequences according to the reference, to allow further processing with GATK (Genome Analysis Toolkit) [10], so as to ensure the quality of reads (all reads are given quality scores and can be dismissed if needed) and perform realignments and recalibrations based on the scores and references, to optimize the output reads and permit the following variance and somatic mutation investigation. Inspection for variance is performed with MuTect [11], which exploits statistical methodologies (Bayesian classifiers) and identifies sites of somatic mutations in paired datasets (tumor vs. normal). To annotate these sites, we use Oncotator [12], which utilizes several databases to link the sites to specific genes. Finally, we perform functional analysis to identify the molecular pathways affected by the specific mutations, and gene prioritization, so as to highlight genes with central role, implicated in diverse and major mechanisms in the Gene Ontology tree. These are performed using BioInfoMiner [13], which combines the StRAnGER2 [14] and GOrevenge [15] algorithms. Figure 1 presents the workflow used here.

Workflow of analysis for the identification of variance and somatic mutations
The complexity of NGS data is high, due to the high amount of information contained in each separate sample (compressed ~10 Gb per sample/20 Gb per patient/~150 Gb for all) and the fact that several distinct parameters need to be adjusted at each step, so as to optimize the performance and the quality of the results (i.e., BWA needs to be adjusted for paired-end sequencing and run in consecutive steps for finding the correct coordinates and generate the final alignment in proper format).
2.2 Analysis of Transcriptomic Data
Transcriptomic analyses among different groups allow the exploration and identification of alterations in gene expression profiles between them. The data used in this section were previously analyzed in [16]. Briefly, the microarray dataset was taken from the Gene Expression Omnibus (GEO) [17, 18], with accession number GDS1375. RNA was isolated from 45 primary melanoma, 18 benign skin nevi, and 7 normal skin tissue specimens and was analyzed for gene expression analysis, using the Affymetrix Hu133A microarray chip containing 22,000 probe sets. Following global normalization, gene expression values across all categories were log transformed, and the mean values of all genes in the normal skin were calculated. Afterwards, the mean gene vector concerning the normal skin categories was subtracted from all replicate vectors of the other two categories. The differentially expressed gene values of the melanoma versus skin, and nevi versus skin, were then analyzed. A false discovery rate for multiple testing adjustment, p-value 0.001 and a 2-fold change threshold for significant differential expression were applied and finally, 1425 unique genes were statistically selected, as being differentially expressed between melanoma and the normal state.
3 Results and Discussion
3.1 Mutational Data Derived from Exome Sequencing
WES data derived from tumor and normal samples were aligned to the human genome, with an average sequence coverage of >100x (number of reads aligning to known reference bases), ideal for achieving the mutational profile required. Overall, the individual samples have depth of coverage >90, with only 3 samples achieving a lower score. Still this lower score is found only in normal samples, which does not affect further analysis, since high coverage is necessary mainly by the tumor samples to overcome endogenous heterogeneity. Table 1 contains the alignment rates and coverage for all samples that were examined. After the processing of individual samples for analysis of variance based on the reference genome, pairs of data from each patient (tumor vs. normal) are jointly analyzed, so as to identify somatic point mutations. Table 2 shows the number of putative sites of somatic mutations, after the MuTect analysis, as well as the count of missense and nonsense mutations for each patient. These mutations affect gene products, by amino acid substitutions or protein truncation, and require further analysis as candidate genetic biomarkers. It is worth noting that the complete workflow for a pair of samples (tumor and normal samples from one patient) needs approximately 35 h running time on a 64 Gb RAM/12 processor cluster server (finally, summarizing the results in ~10 Mb).
In order to discover the molecular pathways affected by the specific mutations, after annotating the mutations to specific genes, we performed functional analysis of the union of affected genes from all the patients (2685 unique genes), which revealed 40 statistically significant biological processes (p-value < 0.05), shown in Table 3.
3.2 Transcriptomic Data
The transcriptomic analysis from [16] revealed 1425 unique differentially expressed genes. Enrichment analysis showed 36 statistically significant biological processes (p-value < 0.05), which are presented in Table 4.
3.3 Data Integration
To facilitate a deeper examination of our datasets, we compared the gene lists from the mutational and transcriptomic analyses. Figure 2 illustrates the total unique and common genes, from the two types of datasets. Only 5 % of the total genes were common between the two sets. Nevertheless, among the highly ranked processes, presented in Tables 3 and 4, cell adhesion, extracellular matrix organization and extracellular matrix disassembly, containing a large number of genes, are found as significantly affected in both cases.

Venn diagram for the significant gene lists from the two analyses
In order to create a feasible disease network for melanoma, we merged the previous results, and carried out an additional functional analysis. This enrichment analysis revealed 45 statistically significant biological processes (p-value < 0.05), presented in Fig. 3, ranked according to their corrected p-values.

Bar plot of significant terms with the number of associated genes (x-axis). Terms are ranked using the corrected p-value. The colors of the genes specify their expression fold change, green -on the left- for under-expressed genes and red -on the right- for over-expressed genes, with neutral indicating somatic mutation
This potential Disease Network revealed several mechanisms with known significance, consistent with melanoma. Enrichment of GO terms, such as epithelial tube branching involved in lung morphogenesis, morphogenesis of an epithelium, epithelial cell differentiation, and regulation of mammary gland epithelial cell proliferation reflects the topological origin of cutaneous melanoma [19, 20]. Furthermore, cell-matrix procedures (organization, adhesion) have been previously reported as significantly altered in tumors [21, 22], as well as lamellipodium assembly, an essential structure for cell migration, which plays an important part in cell invasion and metastasis of cancer [23, 24]. In relation to the ephrin receptor and Rho protein signaling pathways, the Eph receptor tyrosine kinases and their ephrin ligands have specific expression patterns in cancer cells [25], while Rho-like GTPase have been identified as key regulators of epithelial architecture and cell migration, both correlated to cancer development [26, 27].
As expected, the previously discussed significant pathways from Tables 3 and 4 are complemented by the additional data, incorporating an increased number of genes, with considerable implication in melanoma manifestation and progression. Among the significant processes are several previously highlighted by the distinct datasets, but also a number of newly generated, after data integration. Figure 4 indicates the unique and common pathways in each case.

Venn diagram for the significant pathway lists from the two distinct analyses, as well as their integration
4 Conclusions and Future Work
In this study, we sought to export the broader molecular network implicated with cutaneous melanoma. We integrated molecular data of different levels in order to identify the important mechanisms that are involved in this type of cancer. This integration advanced our understanding about the mechanisms implicated with melanoma, by observing the correlation between different sets and levels of data. More importantly, it allowed the manifestation of additional mechanisms previously concealed by the statistical cut-offs, thus enhancing the disease network and our general understanding of the phenomenon.
Our future aim is to expand our current dataset, including data from more patients. Apart from that, we want explore additional methodologies for data integration. Furthermore, our goal is to integrate the molecular data with imaging data from dermoscopy, to improve feature selection and classification techniques, concerning melanoma.
References
The Skin Cancer Foundation. http://www.skincancer.org/
Rossi, C.R., Foletto, M., Vecchiato, A., Alessio, S., Menin, N., Lise, M.: Management of cutaneous melanoma M0: state of the art and trends. Eur. J. Cancer 33(14), 2302–2312 (1997)
Dummer, R., Hoek, K.: Human Melanoma: From Transcriptome to Tumor Biology, Forschungsdatenbank der Universität Zürich (2004–2008)
Winnepenninckx, V., Lazar, V., Michiels, S., Dessen, P., Stas, M., Alonso, S.R., Eggermont., A.M.: Gene expression profiling of primary cutaneous melanoma and clinical outcome. J. Natl. Cancer Inst. 98(7), 472–482 (2006)
Raskin, L., Fullen, D.R., Giordano, T.J., Thomas, D.G., Frohm, M.L., Cha, K.B., Gruber, S.B.: Transcriptome profiling identifies HMGA2 as a biomarker of melanoma progression and prognosis. J. Invest. Dermatol. 133(11), 2585–2592 (2013)
Dutton-Regester, K., Hayward, N.K.: Reviewing the somatic genetics of melanoma: from current to future analytical approaches. Pigm. Cell Melanoma Res. 25(2), 144–154 (2012)
Maglogiannis, I., Goudas, T., Billiris, A., Karanikas, H., Valavanis, I., Papadodima, O., Kontogianni, G., Chatziioannou, A.: Redesigning EHRs and clinical decision support systems for the precision medicine era. In: Proceedings of the 16th International Conference on Engineering Applications of Neural Networks (INNS), p. 14. ACM (2015)
Li, H., Durbin, R.: Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics, Epub (2010). [PMID: 20080505]
Picard tools. http://picard.sourceforge.net
McKenna, A.: The Genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20(9), 1297–1303 (2010)
Cibulskis, K., et al.: Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31(3), 213–219 (2013)
Ramos, A.H., Lichtenstein, L., Gupta, M., Lawrence, M.S., Pugh, T.J., Saksena, G., Getz, G.: Oncotator: cancer variant annotation tool. Hum. Mutat. 36(4), E2423–E2429 (2015)
e-Nios BioInfoMiner tool. https://bioinfominer.com/#/welcome
Pilalis, E.D., Chatziioannou, A.A.: Prioritized functional analysis of biological experiments using resampling and noise control methodologies. In: IEEE 13th International Conference on Bioinformatics and Bioengineering (BIBE), pp. 1–3 (2013). doi:10.1109/BIBE.2013.6701558
Moutselos, K.: GOrevenge: a novel generic reverse engineering method for the identification of critical molecular players, through the use of ontologies. IEEE Trans. Biomed. Eng. 58(12), 3522–3527 (2011)
Moutselos, K., Maglogiannis, I., Chatziioannou, A.: Integration of high-volume molecular and imaging data for composite biomarker discovery in the study of Melanoma. BioMed Res. Int. (2014)
Barrett, T., Troup, D.B., Wilhite, S.E., et al.: NCBI GEO: archive for functional genomics data sets - 10 years on. Nucleic Acids Res. 39(Database issue), D1005–1010 (2011)
Talantov, D., Mazumder, A., Jack, X.Y., Briggs, T., Jiang, Y., Backus, J., Wang, Y.: Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin. Cancer Res. 11(20), 7234–7242 (2005)
Martin-Belmonte, F., Perez-Moreno, M.: Epithelial cell polarity, stem cells and cancer. Nat. Rev. Cancer 12, 23–38 (2012)
Jögi, A., Vaapil, M., Johansson, M., Påhlman, S.: Cancer cell differentiation heterogeneity and aggressive behavior in solid tumors. Upsala J. Med. Sci. 117(2), 217–224 (2012)
Saladi, S.V., et al.: Modulation of extracellular matrix/adhesion molecule expression by BRG1 is associated with increased melanoma invasiveness. Mol. Cancer 22(9), 280 (2010)
Hart, I.R., Birch, M., Marshall, J.F.: Cell adhesion receptor expression during melanoma progression and metastasis. Cancer Metastasis Rev. 10(2), 115–128 (1991)
Machesky, L.M.: Lamellipodia and filopodia in metastasis and invasion. FEBS Lett. 582(14), 2102–2111 (2008)
Kato, T., Kawai, K., Egami, Y., Kakehi, Y., Araki, N.: Rac1-dependent lamellipodial motility in prostate cancer PC-3 cells revealed by optogenetic control of Rac1 activity. PloS one 9(5), e97749 (2014)
Pasquale, E.B.: Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat. Rev. Cancer 10(3), 165–180 (2010)
Ridley, A.J.: Rho proteins and cancer. Breast Cancer Res. Treat. 84(1), 13–19 (2004)
Sander, E.E., Collard, J.G.: Rho-like GTPases: their role in epithelial cell–cell adhesion and invasion. Eur. J. Cancer 35(9), 1302–1308 (1999)
Acknowledgements
This work has been supported by the 12CHN 204 Bilateral Greece-China Research Program of the Hellenic General Secretariat of Research and Technology and the Chinese Ministry of Research and Technology entitled “Personalization of melanoma therapeutic management through the fusion of systems biology and intelligent data mining methodologies-PROMISE,” sponsored by the Program “Competitiveness and Entrepreneurship,” Priority Health of the Peripheral Entrepreneurial Program of Attiki.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 IFIP International Federation for Information Processing
About this paper
Cite this paper
Kontogianni, G., Papadodima, O., Maglogiannis, I., Frangia-Tsivou, K., Chatziioannou, A. (2016). Integrative Bioinformatic Analysis of a Greek Epidemiological Cohort Provides Insight into the Pathogenesis of Primary Cutaneous Melanoma. In: Iliadis, L., Maglogiannis, I. (eds) Artificial Intelligence Applications and Innovations. AIAI 2016. IFIP Advances in Information and Communication Technology, vol 475. Springer, Cham. https://doi.org/10.1007/978-3-319-44944-9_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-44944-9_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-44943-2
Online ISBN: 978-3-319-44944-9
eBook Packages: Computer ScienceComputer Science (R0)