
Abstract
Recent studies from a number of model organisms have indicated chromatin structure and its remodeling as a major contributory agent for aging. Few recent experiments also demonstrate that modulation in the chromatin modifying agents also affect the life span of an organism and even in some cases the change is inherited epigenetically to subsequent generations. Hence, in the present report we discuss the chromatin organization and its changes during aging.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Aging is a complex phenomenon and is a result of many biochemical and molecular changes in the cellular level. The major aging mediated changes in a cell include: reduction in number of mitotic cells; alteration in permeability of membrane; decreased hormone and enzyme secretion; decreased immunity; decreased antioxidant defence with concomitant increase in free radical generation; increased DNA damage (Ashok and Ali 2003). There are many theories to explain aging, such as the gene regulation theory. Recently, change in chromatin structure and epigenetic alterations have also been correlated to aging (Gravina and Vijg 2010). The change in the chromatin structure during aging can be traced back to the observation that chromatin became compact in aged rats with concomitant decline in transcription (Chaturvedi and Kanungo 1985). However, progress in the area of chromatin as a mediator of aging has been relatively slow and was only dependent on few sporadic observations. Few observations of the last decade including role of histone deacetylases in extension of life span in few model organisms has reinforced the link of chromatin to aging.
Here, we describe age dependent changes in the chromatin structure and function. In the first section we give a brief account of the chromatin structure and organization. Then we describe different modes of chromatin remodeling such as ATP-dependent and ATP-independent (post-translational modifications of histones) remodeling. Then we describe different model organisms used for aging study. Finally we describe different changes in the chromatin organization that takes place during aging.
2 The Chromatin Organization
The amount of DNA in a eukaryotic cell is enormous (approximately two meters in case of a diploid human nucleus). Hence, in order to be packaged inside the tiny nucleus, which is approximately 10 μm in diameter, a huge level of folding is needed. To facilitate this, the eukaryotic DNA forms a large macromolecular complex called as chromatin, by associating with almost equal amount of histone and non-histone proteins (Kornberg 1974; Kornberg and Thomas 1974). The fundamental structural unit of chromatin is called nucleosome. Each nucleosome is a complex of a core histone protein octamer, around which 146/147 base pairs of DNA wraps around ~1.7 turns and ~54 bp (10–90 bp) remains as linker DNA. Arrays of nucleosomes appear as beads on a string structure of approximately 11 nm diameter and 5.5 nm in height (Kornberg 1974; Luger et al. 2012). These arrays of nucleosomes form the primary structure of chromatin and popularly named as 10 nm fibre.
The core histone octamer consists of histone H2A, H2B, H3 and H4, each in two copies, which are highly conserved among eukaryotes. These are lysine and arginine rich small basic proteins of molecular weight 11–22 kDa (Luger et al. 1997). The C-terminal regions of each core histone contain five α helical regions. Out of which, the central histone fold motif, comprise of α1 (9–14 a. a.), α2 (~29 a. a.) and α3 (9–14 a. a.). This central histone fold motif is flanked on either side by two other α-helices. These α-helices are connected to each other by short β-loops. The histone fold domains interact with each other to form H2A–H2B and H3–H4 heterodimers. In physiological salt concentrations, two such H3–H4 heterodimers interact with each other by a H3:H3 interaction, to form a tetramer. Similarly a H2A–H2B dimer associates on either side of the H3–H4 tetramer via a H2B:H4 interaction (Luger et al. 1997).
Binding of the core histones to DNA is mostly achieved by weak interactions, such as electrostatic interactions, between the positively charged histone and the DNA phosphate backbone (Luger et al. 1997). The minor groove of the DNA faces towards the histones. There are about 14 such contacts per nucleosome.
The loops of adjacent histones come together and a pair of such loops forms a β-bridge, containing two arginine residues, which interact with the minor groove of DNA. The linker DNA and the nucleosome entry and exit points are bound by H1. The nucleosome containing H1 is called as “chromatosome” which spans ~168 bp (Fletcher and Hansen 1996).
The N-terminal tails of all the core histones, and the C-terminal tails of histone H2A are highly flexible and do not contribute much in the nucleosome formation. They emanate out of nucleosome core, are unstructured and are subject to diverse post-translational modifications (described later). These post-translational modifications act as epigenetic marks for the transcriptional activation and repression. In addition, these tails are responsible for making contact with other nucleosomes and subsequently form higher-order chromatin structure (Li and Reinberg 2011).
These 10 nm fibres undergo next level of compaction to form the 30 nm fibres, in which another six fold condensation of DNA takes place to form a secondary structure. Subsequent formation of the tertiary structure requires interaction between long-distance chromatin fibres by the involvement of histone N-terminal tails (Hansen 2002). The stabilization of higher order chromatin is achieved by the incorporation of linker histone H1 and its variants to the chromatin (Carruthers et al. 1998).
The exact structure of the 30 nm chromatin fibre still remains elusive (van Holde and Zlatanova 1995). However, to explain the conformation several models are proposed (see below). In low ionic strength, the chromatin appears as a beaded fibre, in which beads represent the nucleosomes (Olins and Wright 1973). The 10 nm fibres are wrapped in a left-handed fashion with six nucleosomes per turn to form the 30 nm chromatin structure. Two different models have been proposed to explain the 30 nm chromatin fibre structure; namely the solenoid model and the zigzag model (Robinson et al. 2006). According to solenoid model, the nucleosomes are folded to a simple helix, where the adjacent nucleosomes remain present next to each other in a chromatin fibre. In the zigzag model, the linker DNA is present in a straight path between nucleosomes (Woodcock et al. 1993). Here, two rows of nucleosomes are arranged in a zigzag fashion, due to interaction between alternative nucleosomes. This type of interaction produces a double helical structure (Bednar et al. 1998). Protein-protein cross-linking analysis revealed that from a single stack of 12 nucleosomes, two rows of six nucleosomes were produced as predicted for the zigzag model (Robinson et al. 2006).
The 300 nm fibre, which is the tertiary structure of the chromatin, is formed by folding of the 30 nm fibres. To achieve this folding, the 30 nm fibre assembles into loops, which are also associated with non-histone proteins. These loops are organized being part of scaffold proteins, which maintain its higher order structure (Woodcock and Ghosh 2010). They remain firmly attached to eukaryotic DNA even after its extraction under harsh conditions like high salt, strong detergents and polyelectrolytes. The scaffold is mainly comprised of two proteins i.e. scaffold protein 1 and 2, of M.W. 170 and 135 kDa respectively. The stretches of DNA loops that are associated with the protein scaffold are known as scaffold attachment regions (SARs). The 30 nm chromatin fibre loops are attached to the nuclear matrix via matrix attachment regions (MAR).
The 300 nm chromatin fibres further compact to form 700 nm chromatin fibre during the division phase of cell cycle. The mechanism of 700 nm fibre formation and its exact structure is still unknown. However, the metaphase chromosome is 1400 nm in diameter having two side-by-side 700 nm fibres. Altogether, it can be stated that the structure of 10 nm fibre is well understood and the structure of higher order chromatin structure still remains elusive (Woodcock and Ghosh 2010).
3 The Euchromatin and Heterochromatin: The Classical Domains of Chromatin
Historically, through staining methods, the chromatin was distinguished into a darkly stained heterochromatin and a light stained euchromatin regions. However, it is now established that these domains are differently folded (Grigoryev 2001). Some portions of the chromosomes have a dense, compact structure during interphase, in which genes are repressed, and can be stained with certain dyes. These regions are known as heterochromatin. The regions of chromatin that are relatively unfolded and permissive for gene expression are defined as euchromatin. The euchromatin and heterochromatin domains are interspersed along the chromosome and are demarcated by chromatin barriers known as boundary or insulator elements (Bi and Broach 2001). With recent studies, they have been shown to have differential pattern of post-translational modification in histones and DNA (described later).
4 Chromatin Remodeling
Chromatin becomes inhospitable to nuclear events, such as replication, transcription, recombination and repair, since the DNA remains occluded, being in the form of nucleosomes. This occlusion is reversed by chromatin remodeling. Chromatin remodeling refers to regulated alteration in the chromatin or in the nucleosome structure, making it transiently permissive/occlusive, so as to enhance/obscure binding of factors requiring DNA as template. ATP dependent chromatin remodeling factors and ATP-independent chromatin remodeling factors, with support of each other, mostly bring about the chromatin remodeling (Vaquero et al. 2003).
The ATP-dependent chromatin remodelers are multi-subunit protein complexes of >1 MDa. They utilize ATP and cause disruption in the nucleosome so that the DNA-histone contacts are loosened; mobilize the octamer in cis along a DNA sequence so that a particular DNA sequence becomes accessible; transfer the octamers in trans so that the promoters are freed to be accessed by transcription factors and RNA polymerase; cause looping in DNA; or incorporate histone variants to the nucleosomes. The above mentioned remodeling complexes mostly contain a highly conserved ATPase subunit of helicase superfamily. Depending on the presence of other flanking domains and accessory subunits, the remodeling complexes are further divided into SWI/SNF, ISWI, CHD and INO80 families (Clapier and Cairns 2009).
4.1 SWI/SNF (Switch Defective/Sucrose Non-fermenting) Family
The SWI/SNF group of remodelers (with an approximate size of ~2 MDa) contain an ATPases domain of the SNF2 subfamily and 8–15 other subunits (Vaquero et al. 2003). The C-terminal domain contains a bromodomain for binding to acetylated histones. The SWI/SNF complex was initially identified in yeast. Homologues of yeast SWI2/SNF2 were subsequently identified and characterized from Drosophila (brahma), mouse (mBrm and mBrg) and human (hBRM and BRG1). Similar BRG1 containing homologues have also been identified in rat, Xenopus and chicken (Panigrahi et al. 2003; Vignali et al. 2000). In yeast, there is another SWI/SNF like complex called as RSC (Remodels Structure of Chromatin, size of ~1.5 MDa) (Cairns et al. 1996). The RSC contains four homologues of yeast SWI/SNF components.
The SWI/SNF and related complexes broadly function in reorganization and positioning of nucleosomes in the promoter region for facilitating transcription factor binding. They can cause random positioning of the nucleosomes in a chromatin domain using evenly positioned nucleosomes as substrates. They are also responsible for ejection of nucleosomes and exposing a target DNA site (Dechassa et al. 2010).
4.2 ISWI (Imitation Switch) Group of Chromatin-Remodeling Complexes
Identified first in Drosophila, ISWI-group of remodeling factors has a helicase-ATPase domain, but they lack the bromodomain, unlike the SWI2/SNF2. Instead their C-terminal domain contains a SANT or a SLIDE domain (Clapier and Cairns 2009). Later the ISWI protein was found to be a principal constituent of three ATP-dependent chromatin-remodeling complexes in Drosophila, namely NURF (nucleosome remodelling factor), CHRAC (chromatin accessibility complex) and ACF (ATP-utilizing chromatin assembly and remodeling factor) (Vignali et al. 2000). NURF was purified from Drosophila embryonic extracts as an activity that stimulated transcription factor binding to chromatin promoter. Similarly, CHRAC was purified as an activity that made chromatin accessible to nucleases. ACF, on the other hand, was responsible for nucleosome assembly with the help of histone chaperons (Vignali et al. 2000). ISWI-related complexes were also identified from yeast, Xenopus and human (Dirscherl and Krebs 2004). There is a considerable functional heterogeneity with the ISWI containing complexes. Except NURF, no other ISWI-complex has been shown to disrupt nucleosomes. However, they slide nucleosomes along DNA (cis-transfer) (Dirscherl and Krebs 2004).
4.3 Mi-2/CHD (Chromodomain, Helicase, DNA Binding) Chromatin-Remodeling Factors
These protein complexes were initially identified in mouse (Delmas et al. 1993). They broadly contain an N-terminal chromodomain, usually an essential domain for proteins that interact with and modify chromatin, an ATPase domain (a classical SNF2 group of helicases/ATPases), and a plant homeodomain fingers (a DNA-binding domain). Similar complexes were also identified in yeast, Drosophila, Xenopus, mouse and human (Woodage et al. 1997). In human cells nucleosome remodeling and deacetylase complex, NuRD was identified (Xue et al. 1998). Apart from the regular components of CHD, it also includes histone deacetylase 1 & II. NuRD plays very significant role in cellular processes converging histone deacetylation, DNA methylation and cell-cycle regulation (Fugita et al. 2004).
4.4 INO80 (Inositol Requiring) Factors
These factors are characterized by a split ATPase domain due to a long insertion in the middle. Homologous factors of INO80 exist in yeast (INO80 and SWRI) and human (INO80, SRCAP and TIP60) (Bao and Shen 2007). They demonstrate different kinds of remodeling activities such as nucleosome repositioning, eviction, replacement and exchange of histone with histone variants, mediated by replication independent pathways. They have also role in double strand break repairs of DNA. The TIP60 complex has both histone acetylation and ATPase activity (Morrison et al. 2007).
5 ATP Independent Chromatin Remodeling: Covalent Modification of Histones
The ATP independent chromatin remodelers covalently modify the histone tails or the globular domains. Histones are one of the most conserved eukaryotic proteins and are subject to most diverse number of post-translational modifications such as: acetylation, methylation, phosphorylation, ADP-ribosylation, ubiquitination, biotinylation, sumoylation, etc. (Vaquero et al. 2003). Altogether, they generate accessible or repressible chromosomal domains for further interaction of chromatin complexes.
5.1 Acetylation
Acetylation of histones involves transfer of an acetyl group to lysine residues, present mostly in the N-terminal tails and some core residues of histones. Acetylation of histones is a reversible process. Histone acetyl transferases (HATs) add acetyl groups while histone deacetylases (HDACs) remove it. The acetylated histones become less positive, resulting in decreased affinity for the negatively charged DNA that in turn weakens DNA binding (Hong et al. 1993). This brings about a permissive structure in the chromatin, for binding of different proteins factors to DNA (Lee et al. 1993). Transcriptional activation has been shown mostly synergistic to histone acetylation (Struhl 1998). Further, acetylated histone tails can directly recruit other chromatin interacting machineries, such as bromodomains containing factors. Hence, they act as docking sites for downstream events (Berger 2002; Zeng and Zhou 2002).
In case of vertebrates, all core histones are acetylated at multiple sites. The sites are: H2A (K5, K9); H2B (K5, K12, K15 and K20); H3 (K9, K14, K18 and K23); H4 (K5, K8, K12 and K16), except in yeast, where acetylation sites of H2A, H2B are little different from vertebrates (Roth et al. 2001). There are different HATs, with differential histone substrate specificities. The substrate specificity is accomplished by the interacting partners. The HATs can be broadly divided into type A and type B (Roth et al. 2001). The type B HATs are mostly cytoplasmic and acetylate free histones at sites which are crucial for deposition into nucleosomes. The type A HATs acetylate nucleosome bound histones and broadly belong to three families. The H3 and H4 acetylation is carried by HAT family member Gcn5, while, H4 and H2A are acetylated by the nucleosome acetyl transferase Esa1 (Suka et al. 2001). In vitro all four core histones are acetylated by p300 (Kuo et al. 1996; Ogryzko et al. 1996; Yang et al. 1996).
Histone deacetylation is carried out by histone deacetylases and it results in gene repression. All four core histones are deacetylated by Rpd3 deacetylase (Roth et al. 2001), while, H3 and H2B are deacetylated by Hda1 (Histone deacetylase 1) (Xu et al. 1998). Rpd3 and Hda1 cause deacetylation of larger regions of euchromatin (Vogelauer et al. 2000). Histone deacetylases are also responsible for deacetylation of non-histone proteins, such as p53, tubulin and various transcription factors (Jones and Baylin 2002; Timmermann et al. 2001). Depending on the functional motifs and domains, mammalian histone deacetylases are classified into Class I HDACs (1, 2, 3 and 8), Class II HDACs (4, 5, 6, 7, 9, 10) and class-III HDACs (NAD+ dependent Sir2 family) (de Ruijter et al. 2003; Grozinger et al. 2001). HDAC 11 is shared by both class-I and class-II HDACs. Histone deacetylases have role in turnover of histones, initiation of transcription and repression of active genes.
5.2 Phosphorylation
Phosphorylation of histone occurs in Ser, Thr and Tyr residues. It is also a reversible modification being regulated by kinases and phosphatases (Bannister and Kouzarides 2015). Phosphorylation of H1 and H3 has been correlated with chromosome condensation during mitosis (Hanks et al. 1983; Hirano 2000). Phosphorylation also adds a negative charge to histones. The kinases responsible for phosphorylation of histones can be divided broadly into two categories: (i) those dependent on cyclic nucleotide monophosphates for activation and (ii) those whose activity is independent of cyclic nucleotide monophosphates. However, many other functions of histone phosphorylation still remain elusive.
5.3 ADP Ribosylation
ADP-ribose molecules are transferred either to glutamic acids or arginine residues by poly (ADP-ribosyl) polymerase (Jacobson and Jacobson 1999). Though, all histones are subject to ADP-ribosylation; H2B and H1 are the most preferred substrates (Kappus et al. 1993). ADP-ribosyl transferase transfers single ADP-ribose to arginine or lysine residues of free histones in the cytoplasm. Subsequently, poly-(ADP-ribosyl) polymerase (PARP), carry out the elongation process in nucleus. PARP recognizes and binds to single or double strand DNA breaks and then exposes the DNA strand breaks to proteins involved in repair process (Le Rhun et al. 1998). The ADP-ribosylation is reversed by poly-ADP-ribose-glycohydrolase. Poly ADP-ribosylated histones are part of the relaxed chromatin. Further, it is known that by ADP-ribosylation, the H3K4me3 demethylase is excluded from the chromatin, making the chromatin domain transcriptionally active (Li et al. 2014).
5.4 Ubiquitination
Ubiquitin is a 76-amino acid protein (Conaway et al. 2002). Ubiquitination is another reversible modification, which occurs at lysines residues of histone H2A and H2B during cell cycle. H3 and H1 are also reported to be modified by the same mechanism (Belz et al. 2002). Monoubiquitination is a mark of both transcriptional activation and repression depending on the position and histone subtype. Monoubiquitination of H2BK123 in yeast is crucial for methylation of H3K4, a mark of transcriptionally active chromatin (Sun and Allis 2002). Similarly, monoubiquitination of H2AK119 is a mark if transcriptionally inactive chromatin. In vitro, polyubiquitination of histone H2A, H2B and H3 are targets for degradation (Jentsch and Schlenker 1995).
5.5 Sumoylation
Small Ubiquitin related modifiers (SUMO) is also involved in posttranslational modification of histones on lysine residues (Melchior 2000; Nathan et al. 2003; Robzyk et al. 2000; Shiio and Eisenman 2003). SUMO shares only 18 % identity with Ubiquitin group. Out of the four histones, histone H4 is efficiently modified by SUMO-1 or SUMO-3, both in vitro as well as in vivo. Acetylated histone H4 usually gets sumoylated. It leads to decreased acetylation of histone H3 due to recruitment of HDACs and increase in HP1 binding. Hence, sumoylation has a role in repressing transcribing chromatin (Girdwood et al. 2003). It has been hypothesized that probably sumoylation initiates attenuation followed by gene repression.
5.6 Lysine Propionylation and Butyrylation
Histones are subject to propionylation at K5, K8 and K12 of H4 and lysine butyrylation at K5 and K12 of histone H4 (Nathan et al. 2003). These sites are also known to be acetylated (Tanaka et al. 2004). Probably these two new types of modification create novel docking sites for downstream recruitment of other complexes which are yet to be elucidated.
5.7 Deimination
In this type of modification, an arginine or monomethyl arginine residue of the histone is converted to citruline (Cuthbert et al. 2004). In mammals, this conversion is catalysed by peptidyl deiminase 4 (PADI4) and this enzyme is known as arginine demethylase. The resultant change of the citruline instead of arginine reduces in the histones affects the positive charge of the histones and it also abolishes the respective docking site.
5.8 Methylation and Demethylation
Histone H3, H4 and H1 can be methylated on their arginine (R) and lysine (K) residues (DeLange et al. 1969; Gershey et al. 1969; Murray 1964; Patterson and Davies 1969). Depending on the number of methyl groups attached to ε amino group, lysines can be mono, di or tri methylated. Similarly, arginines can be mono or dimethylated. Lysine methylation can have mixed effects. It depends on the position of lysine residue which is methylated and the levels of methylation (mono, di or trimethylated). Methylation at K4, K36 and K79 of histone H3 are marks of transcriptionally active chromatin, while methylation at K9, K27 of H3 and K20 of H4 are marks of transcriptionally inactive chromatin (Cao et al. 2002; Peters et al. 2003; Rice et al. 2003; Schotta et al. 2004). The lysine methylation is reversibly regulated by histone methyl transferases and demethylases. Histone methyl transferases (HMTs) methylate histone lysine and arginine residues. They have been broadly classified into three protein families: the PRMT family methylates arginine residues (Bedford and Richard 2005; Zhang and Reinberg 2001), while the SET domain containing family and the Dot1/DOTL families methylate lysine residues (Bannister et al. 2002; Kouzarides 2002; Lachner et al. 2003). The SET1 group is specific for H3K4 (Ringrose and Paro 2004; Smith et al. 2004), the SUV 39 group for H3K9 (O’Carroll et al. 2000; Rea et al. 2000; Tachibana et al. 2001; Yang et al. 2002), the SET2 group for H3K36 (Peters et al. 2001) while ASH1 shows broad specificity and methylates H3K4, H3K9 and H4K20. The EZH family methylate K27 and K9 of histone H3 (Czermin et al. 2002; Muller et al. 2002), Dot1 containing proteins (Singer et al. 1998) methylates H3K79 of the core domain (Feng et al. 2002; Lacoste et al. 2002; Ng et al. 2002; van Leeuwen et al. 2002). However, in many of these methylases, cross-reactivity is also reported.
Unlike acetylation, in methylation, there is no major change in total charge on histones. Rather specific lysine residues, after being differentially methylated (mono, di or tri), act as docking sites for further recruitment of other interacting machineries. It finally results in generation of transcriptionally permissive/occlusive chromatin template (Strahl and Allis 2000; Turner 2000). For example, the chromodomain containing protein HP1 (heterochromatin protein 1) is recruited to methylated histone H3K9 and generates a heterochromatin domain (Bannister et al. 2001; Nakayama et al. 2001). The heterochromatinization spreads by self-association of many HP1 (Nielsen et al. 2002). Antagonistically, chd1, the chromodomain containing protein (a component of SAGA and HAT complex) is recruited to methylated H3 K4, and it further recruits SAGA and HAT complexes and ultimately generates a euchromatic chromatin domain (Pray-Grant et al. 2005).
5.9 Lysine Methylation in Transcriptional Silencing
Methylation of K9 and K27 of H3 and K20 of H4 are marks of transcriptionally silenced chromatin domains (Bannister and Kouzarides 2015). Methylation of H3K9 is a deterministic mark of constitutive heterochromatin and also marks chromatin domains for transcriptional silencing. The differential role is played by the fact that euchromatic genes are regulated being dimethylated at H3K9 and the heterochromatic silencing takes place by generating trimethylated H3K9. However, few deviations are also available (Vakoc et al. 2005). Similarly, H3K27 methylation is also a mark of transcriptional silencing and X-chromosome inactivation.
There are reports that methylation of H9 of H3 (H3K9me) is a prerequisite for DNA methylation (Jackson et al. 2002; Lehnertz et al. 2003; Tamaru and Selker 2001), as binding of HP1 to H3K9me recruits DNA methyl transferases. It has been further demonstrated that E2H2 complex, which methylates H3K27, also interacts with DNA methyl transferases (Vire et al. 2006). However, results supporting the opposite hypothesis have also been documented. There is significant reduction in H3K9 methylation marks in DNA methyl transferase deficient cells (Sarraf and Stancheva 2004).
H4K20 methylation also correlates with the condensed regions of chromatin. In vitro its methylation inhibits of H4K16 acetylation, which is a mark of transcriptionally active chromatin domain. The H4K20me is also an epigenetic mark of repression and generally goes synergistically to H3K9 methylation (Zhang and Reinberg 2001).
5.10 Lysine Methylation and Active Transcription
5.10.1 Methylation of H3K4
The complex associated with setI (COMPASS) in association with elongating RNA Polymerase II, methylates H3K4 (Briggs et al. 2001; Daser and Rabbitts 2004; Gerber and Shilatifard 2003; Krogan et al. 2002; Miller et al. 2001). The COMPASS of the MLL complex are classically known as generators of euchromatin marks (Roguev et al. 2001). Components of COMPASS generate trimethyl K4 of histone H3 (H3K4me3) marks (Schneider et al. 2005). Actively transcribing genes are enriched with both dimethyl and trimethyl H3K4 marks. While, the dimethyl modifications mostly restricted to the coding regions, the trimethyl modification localize specifically to the regulatory regions of genes (Bernstein et al. 2005). WDR5 and Chd1 are recruited to the methylated H3K4, and they acetylate other lysine residues like H3K14 and K9. The K9 acetylation mark is antagonistic to K9 methylation mark and thus the respective chromatin domain remains in an activated state (Timmers and Tora 2005). Further, the WDR5 binds to H3K4me2 and is responsible for maintaining global trimethylated H3K4 condition (Wysocka et al. 2005).
5.10.2 Methylation of H3K36
Set2 was initially identified in yeast to have HMTase activity specific to H3K36 (Strahl et al. 2002). Set2 was speculated to have role in transcriptional elongation as was reported to associate with phosphorylated RNA Polymerase II (Krogan et al. 2003). It is thus hypothesized that recruitment of Set2 along with RNA Pol II is crucial for establishing K36 methylation on chromatin (Gerber and Shilatifard 2003, Hampsey and Reinberg 2003).
5.10.3 Methylation of H3K79
The nonset domain containing protein dot1 (disrupter of telomeric silencing 1) methylates the core domain of nucleosomal histone H3. It is further seen that 90 % of the nucleosomal histone H3K79 remains methylated (van Leeuwen et al. 2002). The H3K79me mark prevents binding of silent domain Sir Proteins and maintains the chromatin in euchromatic state and prevents further heterochromatinization.
5.10.4 Histone Lysine Demethylation
Histone methylation was considered to be a relatively permanent modification since, the histone demethylases were not known. However, subsequently the histone demethylases have been identified and characterized, at least for quite a few methylated residues. Lysine specific demethylase-1 (LSD1) was shown to be specific for H3K4me2 (Shi et al. 2004). LSD1 is a component of Co-REST co-repressor complexes (Hakimi et al. 2002; Shi et al. 2003; You et al. 2001). Demethylation by LSD1 generates unmethylated histones, formaldehyde and H2O2 and in the process FAD+ is reduced to FADH2, which is then reoxidised by (O). The (O) is reduced to form H2O2. However, LSD1 shifts its specificity from H3K4me2 to H3K9me2, and acts as a H3K9 demethylase and transcriptional activator in the presence of Androgen receptor (AR) (Metzger et al. 2006).
Another group of proteins called Jumonji (JmjC) proteins are shown to have histone dimethylase activity (Tsukada et al. 2006). JHMD1/Fbx11 (JmjC domain containing histone demethylase1, alias Fax11) demethylates mono and di meH3K36. Similarly, JHDM2 demethylates H3K9me2 (Yamane et al. 2006). Recently JMJD 2A-2D groups are identified. They reverse trimethylation, specifically H3K9me3 and H3K36me3 (Whetstine et al. 2006). Since, H3K9me3 and H3K36me3 are total antagonists: as the former is a mark of transcriptionally inactive region and the later is a mark of region engaged in transcriptional elongation. The dual site specificity of the JMJD2A/C may suggest a coordinated role in regulating H3K9/K36 trimethylation for gene regulation (Carrozza et al. 2005).
5.10.5 Histone Arginine Methylation
Arginine methylation is mainly linked to active transcription. Protein arginine methyl transferases (PRMTs) transfers a methyl group to arginine residues (Gary and Clarke 1998). Arginine methylation occurs in R2, R17 and R26 of histone H3 and R3 of histone H4. Arginines can be mono- or dimethylated. Further, the dimethylation can be symmetric or asymmetric. The arginine methyl transferases (PRMT) which methylate Arg residues have been divided into two groups. The type-I enzyme catalyse the formation of -monomethyl and asymmetric dimethyl arginine residues, whereas the type-II enzyme catalyzes the formation of monomethyl and symmetric dimethyl arginines (Zhang and Reinberg 2001).
5.10.6 Reversal of Arginine Methylation
Monomethylated and unmethylated arginine can be converted to citrulline by arginine deiminase, PADI4 (Cuthbert et al. 2004). PADI4 can deiminate unmodified arginine and monomethyl (but not dimethyl) arginine. It is a Ca+2 dependent enzyme that acts as a repressor of transcription, in the pS2 promoter. It demethylates H3 and H4 arginines (mono or unmethylate) by deimination, at positions R2, R8, R17 and R26 of histone H3 in vitro and R2, R8 and R17 in vivo. Similarly, JMJD6 demethylates H3R2me2 and H4R3me2 in vitro and in vivo (Chang et al. 2007).
5.10.7 DNA Methylation
Methylation also occurs in few nucleotides of DNA and is often synergistic or antagonistic to few histone methylations (described earlier). 5-methyl cytosine and subsequent guanine (5meCpG) dinucleotides are mostly marks of gene silencing and also confer epigenetic memory. DNA methylation is regulated by de novo DNA methyl transferases and maintenance methyl transferases (Marchal and Miotto 2015). The first group of methyl transferases, DNMT3a and DNMT3b, methylate one strand of the newly synthesized DNA and convert them to hemimethylated DNA. The hemimethylated DNA is converted to a methylated DNA by maintenance methylase, DNMT1. However, DNMT3a and DNMT3b have been shown to also act on hemimethylated DNA (Marchal and Miotto 2015). Few recent reports have also demonstrated methylation outside the CpG (Yang et al. 2013). The human genome has approximately 30 million CpG. Dense clustering of these CpG in some regions of the genome is observed and these regions are known as CpG islands. Presence of 5meCpG islands in the promoter region of a gene is a mark of silenced gene. 5meCpG in the coding region of a gene is also observed, however, its mode of regulation is yet to be properly understood (Shenker and Flanagan 2011). DNA demethylases were not known until recently and hence DNA methylation was believed to be a relatively permanent mark. Now, formation of 5-hydroxymethyl cytosine (5-hmeC) is believed to be an intermediate in DNA demethylation process (Pfaffeneder et al. 2011). Oxidation of 5-meC generates 5-hmeC and then to several intermediates which are observed (Jurkowski and Jeltsch 2011). These reactions are catalysed by TET (ten-eleven translocations) enzyme family. Embryonic stem cells have higher levels of 5-hmC and it decreases upon differentiation (Xu et al. 2011). However, few other methods are also proposed for demethylation of DNA. They include non-replacement of methylation marks in the daughter cells during division, removal of methyl group by DNA glycosylase and deamination of cytosine or oxidation.
5.10.8 The Histone Code and Cross-Talk in Histone Modification
Many of the histone modifications act as docking sites and cause further recruitment of protein complexes that bring about broadly two effects: a permissive chromatin domain for binding of factors; or an occlusive chromatin domain refractory for factor binding. Accordingly, a group of modifications coexist and work synergistically but are not compatible with others group of modifications (Jenuwein and Allis 2001). Say, for example, when H3K4 is methylated, there is synergistic acetylation of H3K14 and phosphorylation of H3S10. All are involved in transcriptional activation, and all of them are antagonistic to the inhibitory H3K9 methylation. Further, the complexity increases as lysines can be mono, di or tri methylated. Accordingly, they encode different signals for activation. Taken together, a language of histone modification can be created with the available information, which was initially framed as histone code (Strahl and Allis 2000). This code was then supported by yet another hypothesis that the simplest code can be binary (Jenuwein and Allis 2001). For example, H3K9 and H3K4 cannot be methylated simultaneously in the same chromatin domain. Likewise, H4K20 methylation and H4K16 acetylation are antagonistic. Distinct histone modifications, on one or more tails, act sequentially to form a “histone code”, that is read by chromo and bromodomain containing proteins. This creates specific combinations of modifications on a local domain. All together they bring about formation of the euchromatic and heterochromatic domains of chromatin. A detailed account of the different post-translational modification and their cross-talk is represented in Fig. 1.

Histone post-translational modification and their cross-talk: The N- and C-terminal core histone tails are represented with a. a. residues that are subject to different modifications. Here, the proteolytic cleavage is also included. Further, few deterministic cross-talks between the modified docking sites are also represented
5.10.9 The Proteolytic Processing of Histones
One permanent way of removal of the histone modification is by clipping the histone tails. Proteolysis of histones, though rare, is believed to be another mode of irreversible post-translational modification (Allis et al. 1980). The N-terminal/C-terminal tails of histones are more susceptible for the proteolysis, when histones exist in the form of chromatin. Proteolysis of histones emerged as a problem for chromatin isolators (Brandt et al. 1975). However, later the site specific histone proteolysis has been linked to aging and chromatin structure modulation (see below).
In calf thymus, chromatin-bound proteolytic activity which was specific to H2A was demonstrated (Eickbush et al. 1988). The cleaved product, which lacked 15 a. a. from the C-terminus, was named as cH2A. The cleavage site was between Val-114 and Leu-115. (Okawa et al. 2003). Two truncated forms of H2A have been purified from the nuclei of acute myeloid leukemia OCI/AML1a cells (Okawa et al. 2003). However, the enzyme catalysing this cleavage is not known. Proteolysis of histones has also been proposed during transformation of spermatids into spermatozoa (spermeogenesis), at the terminal stages of spermatogenesis (Marushige et al. 1976). During the process, chromosomal DNA becomes tightly packaged as a result of proteolytic replacement (Marushige and Marushige 1983) of somatic type histones, by protamines. The mechanism of histones displacement requires their proteolysis for removal. A protease activity associated with mouse testicular chromatin has also been observed, which might be involved in proteolytic removal of somatic histones during spermatogenesis (Faulkner and Bhatnagar 1987). Further, this specific proteolysis was absent in other tissues like thymus and liver suggesting tissue specific functional regulation.
Selective and site specific proteolysis of a specific histone subtype has also been observed during viral infection. There is histone H3 degradation in mammalian kidney cells (BHK cells) infected with Foot-and-Mouth Disease Virus (FMDV) (Grigera and Tisminetzky 1984). Further, it is demonstrated that the H3 degradation is catalysed by the FMDV 3C protease and cleavage site has been mapped between Leu-20 and Ala-21, from the N-terminal end (Falk et al. 1990). As the processed H3 lacks most of the N-terminal acetylation sites, it would shut off/reduce the host cellular transcription (Tesar and Marquardt 1990).
Proteolytic processing of histone H1 and H3 has been observed as a physiologically and developmentally regulated event in Tetrahymena nuclei (Allis et al. 1984). Histone H3 is cleaved at multiple site spanning between a.a. 21–27 by Cathepsin L during mouse embryonic stem cell differentiation (Duncan et al. 2008). The protease Cathepsin L is extensively characterized. The H3 cleavage by Cathepsin L is regulated by specific covalent modification code of histone H3. Recently, in chicken liver tissue, glutamate dehydrogenase was identified as a histone H3 N-terminus specific protease (Purohit et al. 2013). A yet to be identified histone H2A specific protease is also characterized from chicken liver (Panda et al. 2013). In summary, in recent years histone specific proteolytic cleavage and histone subtype non-specific degradation have also been observed simultaneously, suggesting that histone-specific and histone non-specific proteases exist in nature and they have evolved along with histones. Hence, it is recently opined to distinguish these proteases into broadly two groups: 1. histone proteases with broad spectrum specificity mostly function for turnover of histones. 2. Histone proteases with site specificity have role in modulating chromatin structure and function by modulating epigenetic marks (Dhaenens et al. 2015).
6 Change in Chromatin During Aging
With the preliminary understanding on the chromatin organization and function, in the following section we intend to describe the age dependent change in the chromatin organization and function. Since, aging is naturally a slow process to score, at least in a number of higher organisms; simple models are developed to study aging. So in the first part of the following section, we describe different suitable model systems which are presently being studied to understand the chromatin structure and aging. In the subsequent section we will describe different changes in the chromatin organization and function that take place during aging.
7 Organismic Models for Studying Chromatin and Aging
7.1 Yeast
Due to the ease in genetic manipulation, short life cycle and conservation of mechanisms of aging across the eukaryotes, the budding yeast Saccharomyces cerevisiae is a growing model for aging study (Wood and Helfand 2013). One of the measurement parameters for aging is the replicative life span in budding yeast or in other words the number of daughter cells a budding mother cell produces before it reaches non-dividing stage. It has been observed convincingly that there is loss of transcriptional silencing with concomitant decrease in chromatin compaction in budding yeast during the process of aging (Smeal et al. 1996). In budding yeast, three regions of the genome remain silent: viz. the mating type switching locus (MAT locus), the ribosomal DNA locus (rDNA) and the telomere proximal regions. In aged yeast, there is loss of silencing of the MAT locus leading to co-expression of both mating type genes, causing insensitivity to pheromones and increased sterility in aged yeast. Further, there is movement of silencing proteins such as HDAC (NAD+ dependent histone deacetylase) and silencing information regulator 2 (Sir2) away from the telomeric region in the aged yeast (Kennedy et al. 1997). Also, in the aged yeast, there is loss in silencing of the rDNA region with concomitant rise in recombination and formation of extra ribosomal chromosomal circles and increase in genomic instability. These events can be monitored with limiting replicative life spans. It is further seen in yeast model that deletion of Sir2 reduces replicative life spans, while introduction of an extra copy of Sir2 gene increases the same (Etzioni et al. 1999). Hence, from these observations, it can be concluded from yeast model that chromatin of the silent regions remains in more relaxed state in aged yeast. Sir2 is known to deacetylate H4K16Ac (Imai et al. 2000). Further, there is an age dependent decrease in Sir2 proteins in yeast leading to increase in H4K16Ac mark in global level (Dang et al. 2009). The H4K16Ac is a unique epigenetic mark which inhibits formation of 30 nm fibres and subsequent folding of the chromatin into higher order. Thus, age dependent increase in H4K16Ac leads to formation of open chromatin structures globally (Shogren-Knaak et al. 2006).
There is also change in the gene expression pattern during aging in yeast (Wood and Helfand 2013). Some of the change in gene expression could be due to elevated stress responses and induction in repair genes during aging. However, a significant amount of change in the gene expression could also be due to altered chromatin structure during aging. It is observed that there is increased transcription of the core histones with aging. On the other hand, short-lived yeast mutants deficient in the ability to acetylate H3K56Ac have decreased level of core histone transcript in aged cells, which is in turn similar to that of telomerase deficient cells (Lesur and Campbell 2004). H3K56Ac is enriched in histone gene promoters for transcription. Conversely, elevation in the histone transcript levels in aged yeast does not correlate with increase in bulk histones in the aged yeast. In fact, there is decrease in bulk histones in aged yeast cells (Feser et al. 2010). This could be due to the feedback inhibition of the histone transcript translation. Further, by ChIP analysis it has been revealed that there is reduced histone occupancy in many regions of the chromatin including the telomere proximal region, the rDNA region and the MAT locus in the aged yeast (Wood and Helfand 2013). The loss of histone from the chromatin of aged yeast leads to the formation of a more open chromatin and in turn increases transcription of some genes. On the other hand, lower concentrations of the synthesized histones in the aged yeast leads to decreased pool of free histones and also alters epigenetic marks of the histones. In another line of evidences, it has been found that aged yeast cells have decreased polyamine synthesis (amines are required for cell proliferation). Yeast cells treated with spermidine showed hypoacetylation of H3K9, K14, K18 and H4K16 with concomitant decrease in HAT activity, specific for these sites and increase in replicating life span (Eisenberg et al. 2009). However, it was in contrast to the hyperacetylation of the H3K56 sites suggesting again the site specific role of diverse histone acetylations in modulation of aging in yeast model.
7.2 Drosophila
Similar to the change in the acetylation at multiple sites of histone H3 and H4, the marks of active chromatin, from aged yeast, a decline in few methylation sites (which are mark of active chromatin) have also been reported from aged Drosophila models. There is decrease in H3K4me3 and H3K36me3 marks with age in Drosophila (Wood and Helfand 2013). However, there is also less enrichment in the H3K9me3, and Heterochromatin protein1, HP1 (mark of inactive chromatin) in the pericentric heterochromatin. These flies with decreased HP1 expression also exhibited increased levels of rRNA transcripts. Recently it has been reported that flies with decreased HP1 expression exhibited shorter lifespan whereas in flies where HP1 was overexpressed, there was increase in the lifespan (Larson et al. 2012).
7.3 Caenorhabditis elegans
In C. elegans, a number of histone modifications have been recently correlated with the life span of the organism. The methylation marks of K4, K9 and K27 of H3 are extensively studied in the worm (Padilla et al. 2014). It is found that disruption of the H3K4 methyl transferase complex (ASH-2), leads to increase in the lifespan of the worm, while disruption of the H3K4 demethylase (RBR-2) leads to decrease of the same. It has been further shown that this increase in life span is inherited up to certain generations (Greer et al. 2011). Similar observations are also reported for other modifications sites of histone H3. An aging dependent decline in the H3K27me3 is observed in the worm with concomitant increased activity of the respective histone demethylase (UTX-1). It has been further seen that disruption of UTX-1 results in increased levels of H3K27me3 marks in the worm genome and there is increase in the life span (Maures et al. 2011). Synergistically, disruption of the LSD1 complex that acts as a demethylase for H3K4me and H3K9me sites also results in increased life span in the worm (McColl et al. 2008). However, it is not clear from these observations, why there is an increase in life span on hypermethylation of activation marks such as H3K4 as well as repression marks such as H3K9 and H3K27. It can be suggested that probably gene specific transcription plays a more important role during aging than the global level change in the transcription of an organism which can also be tissue specific.
7.4 Mammals
Mouse, rats and few human diseases are also used as good models to study age related changes in chromatin. Analysis of the 30 nm fibres and Micrococcal nuclease (MNase) digestion of the nuclei have indicated that there is irregular positioning of the nucleosomes in the chromatin of aged fibroblasts. It suggests that there is loosening of the chromatin structure with age (Macieira-Coelho and Puvion-Dutilleul 1989). There is also decrease in heterochromatic regions like less efficient inactivation of the X-chromosomes in aged mice (Cattanach 1974).
Genetic disease such as progeria in mammals mimics the aging process and pose as a good model for studying aging (Sedivy et al. 2008). The disease is caused due to mutation in Lamin A gene which forms a part of the nuclear envelope (Oberdoerffer and Sinclair 2007). Mutation of the Lamin A gene by altered splicing, generates a truncated lamin A protein, termed as progerin (Feser and Tyler 2011). Accumulation of lamin A in the nuclear envelope causes a number of downstream effects. There is decline in methylation marks for heterochromatin, like H3K9me3 and H3K27me3 levels synergistic to decline in HP1 and EZH2 (H3K27 methyl transferase). It results in increased transcription from pericentric heterochromatin, shortening of telomeric regions and disruption of nuclear architecture. Progerin expresses at very basal level in young and old mammals. Overexpression of progerin results in all these change in the histone modifications status, described as above and disorganization of the nuclear architecture. Even, by blocking the cryptic splicing site and correcting the splicing defect, the abnormal cell can be reverted back to normal and delay aging process (Feser and Tyler 2011).
It has been shown that healthy aged cells have decreased levels of NURD complex (Meshorer and Gruenbaum 2009). Progeric cells also show decreased level of NURD complex and concomitant loss of HP1 and H3K9me3 along with severe DNA damage (Feser and Tyler 2011). Hence, these systems also pose a good model to study NURD mediated epigenetic signalling for aging.
8 Change in Chromatin Structure and Function During Aging
8.1 Change in Nuclear Architecture During Aging
As discussed earlier, in yeast, the heterochromatic regions mainly silence the mating type locus and stabilize the rRNA genes and telomere regions by preventing them from recombination. As the rDNA region is repetitive, it is also prone to recombination. In yeast, the rRNA region is stabilized and silenced by REST (regulator of nucleolar silencing and telomere exit) complex (Oberdoerffer and Sinclair 2007). This complex is a Sir2 containing complex. Sir2 is a major histone deacetylase responsible for deacetylating histone H4K16Ac. The Sir2 interacts with Sir3 and Sir4 not only in the rDNA region but also in the MAT and the telomeric regions and is regulated by them (Kaeberlein et al. 1999). In yeast, the highly repetitive rDNA region is prone to recombination. Recombination mediated excision in these regions generates extra-chromosomal circles. During subsequent cycles these extra-chromosomal circle keep on accumulating in the nucleolus and cause cell death. Hence, there is a need to silence the recombining rDNA region to increase genomic stability and delay aging, which is done by the Sir2 complex. A truncated Sir4 protein, no longer interacts with the telomeric region. Instead, more Sir2 and Sir3 are targeted to the nucleolus. It results in extension in life span of the yeast (Straight et al. 1999).
Similar observations are also obtained from human progeroid disease, Werner Syndrome (WS) (Oberdoerffer and Sinclair 2007). In this disease, there is a mutation in the DNA helicase gene. This mutation also causes genomic instability by extra-recombination in the rDNA region and results in early aging. In other similar diseases such as HGPS (Hutchinson-Gilford Progeria Syndrome), there is a mutation in the Lamin A gene which, encodes a protein of the nuclear membrane, as described earlier (Oberdoerffer and Sinclair 2007). It disturbs nuclear membrane architecture with concomitant loss of HP1 proteins. HP1 is very much essential for maintenance of the integrity of the constitutive heterochromatic region as it binds to methylated H3K9. Loss of HP1 leads to disturbance in the heterochromatic regions. Further findings have shown that a similar splice variant of lamin A also accumulates in normal aged individuals indicating similar events in the normal aging and disease induced aging processes (Scaffidi and Misteli 2006). Similarly, in the AT (ataxia telangiectasia) disease also there is mutation in the ATM gene which is a component of the DNA repair cascade important for telomere maintenance. Defective ATM products also disturb telomere and nuclear matrix interaction and enhance aging (Greenwell et al. 1995).
Age dependent changes in the heterochromatic regions, such as centromeric region are also reported. The pericentric-heterochromatin region around the centromere becomes transcriptionally more active in aged cardiac tissue (Gaubatz and Cutler 1990). There are further supporting observations hypothesizing that change in the perinuclear architecture contributes to aging in a number of organisms (Oberdoerffer and Sinclair 2007). Few recent observations have shown that there is a notable increase in the total facultative heterochromatin domains in the senescent cells (Adams 2007). By this process there is large scale reorganization of the heterochromatic regions in an age dependent manner.
8.2 Age Dependent Change in Core Histone Expression and Deposition into Nucleosomes
An age dependent change in expression of core histone is recently reported in many organisms such as yeast and mammals. There is reduction in the core histone expression during replicative aging in yeast with concomitant decrease in histone occupancy (Hu et al. 2014). It leads to aberrant up-regulation of associated genes and also causes genomic instability. Further, in yeast models, deletion of anti-silencing function-1 and CAF-1, (the central chaperone complex required for deposition and removal of histones from chromatin) causes drastic reduction in its life span (Feser et al. 2010). It has been seen that though there is significant increase in the histone transcripts during aging, there is global decrease in the chromatin bound core histones during aging. Supply of extra core histone or ectopic expression of histone H3 and H4 and not H2A and H2B increases the replicative life span in yeast. Hence, it is concluded that expression of H3 and H4 dimer promotes nucleosome deposition and transforms again to a more uncompact form and thereby decrease genomic instability and increase life span. Though, to the best of our knowledge, similar experimental observations are limiting from mammalian cells, it can be argued that similar process can also be applicable to mammalian systems, as similar decline in histone expression is also observed in mammalian cells too.
8.3 Change in DNA Methylation
In vertebrates, transcriptionally silent regions are generally marked by the presence of 5-methyl cytosines in the CpG dinucleotide sites (described earlier). There is reduction in the 5meC in aged human cells. Further studies have indicated that there is decrease in the 5meC in the CpGs present outside the promoter regions in different human tissues (Benayoun et al. 2015). Conversely, there is an increase in 5meC in the promoter region of aged tissues of human and mice. It is further hypothesized that the methylation in the promoter regions of development related genes probably contribute to the aging mediated mis-regulation of gene expression. Recent reports have suggested a link between human age and 5meC status of DNA (Zou et al. 2014). The stem cells (embryonic and pluripotent stem cells) are estimated to be ageless in correlation to the DNA methylation age. Similarly, sperms and ovum have younger DNA methylation ages compared to respected differentiated tissues from the same individuals. Further, a comparison of the DNA methylation status of different tissues can also be useful for predicting the health of various tissues. For example, in obese individuals, there is increased DNA methylation age of liver than other tissues of the same individual. Similarly, individuals with Down’s syndromes have increased methylation age of blood cells compared to normal individuals with similar biological age.
Similar observations are also seen in model organisms for aging study. In Drosophila, overexpression of dDnmt2 (DNA methyl transferase) gene increases longevity, whereas deletion of the gene generates short lived flies (Benayoun et al. 2015). Although, the mechanism of methylation mediated modulation of aging is still unclear, the results obtained from Drosophila and human samples pose a relationship between DNA methylation and aging and suggests that DNA methylation could be used as a general biomarker for aging.
8.4 Changes in Histone Methylation
As discussed earlier, methylation of core histones at specific sites, mark either the active or repressive chromatin. Further, histone methylation is reversibly regulated by histone methyl transferases and demethylases. With aging, there is global loss of heterochromatin domains with concomitant redistribution of heterochromatin silencing proteins (Tsurumi and Li 2012). Increase in H3K27me3 by decreasing expression of H3K27me3 demethylase results in increase in life span in C. elegans (Ni et al. 2012). However, the results are contradictory from different organisms. In Drosophila, reduction in expression of H3K27me3 methyl transferase with concomitant reduction in H3K27me3 increases life span (Ni et al. 2012). On the contrary, in muscle stem cells of old mice, there is increase in gross H3K27me3 (Baumgart et al. 2015; Liu et al. 2013). The contradictory increase or decrease in methylation status of H3K27 further suggests the fact that the methylation signals are tissue and organism dependent.
Age dependent redistribution of H3K4me (a mark of active chromatin) is also observed (Shah et al. 2013). Spreading and redistribution of H3K4me3 occurs in aging fibroblast of humans and hematopoietic stem cells of mice. Further, modulators of H3K4me also influence longevity. It is observed that knocking down the main component of the H3K4 methyl transferase complex increased lifespan in C. elegans. On the contrary, when the respective demethylase gene was knocked down, it resulted in shortened lifespan (Greer et al. 2011). Further, overexpression of the H3K4 demethylase in the germinal cells increased lifespan. Similar observations are also obtained from other histone H3 amino acid residues which act as site for methylation. Mutation of the H3K36 demethylase gene causes expansion in replicative age in yeast. Synergistically, a mutation the K36 residue in H3 of yeast, which makes it refractory to be methylated, shortens life span (Sen et al. 2015). Further, in C. elegans, knockdown of the H3K36me3 methyl transferase gene also shortens life span of the worm (Pu et al. 2015).
8.5 Changes in Histone Acetylation
The acetylation pattern of core histones has been reported to change during normal aging in a number of organisms (Benayoun et al. 2015). In yeast models, during replicative aging, there is bulk level decrease in H3K56Ac, while there is increase in H4K16Ac (Dang et al. 2009). On the contrary, there is age dependent decrease in H4K16Ac in mouse and progeroid models (Krishnan et al. 2011). In aged mice also there is a lack of transcription mediated upregulation in H4K12Ac, which is a mark of transcriptional elongation.
Concomitant to the age dependent increase in H4K16Ac, the enzyme specific for deacetylating the H4K16Ac, the Sir2 also modulates life span in yeast. Increasing dose of Sir2 (Sirtuin silent information regulator 2) or inducer of Sir2 such as Resveratrol, extends life span in yeast and other model systems (Kaeberlein et al. 1999). Similarly, orthologues of Sir2 such as SIRT6 in mice deacetylates H3K9Ac and H3K56Ac. Deficiency of SIRT6 generates progeroid like phenotypes in mice, hypothesizing that it has role in aging. On the contrary, overexpression of sirt6 increases longevity (Kanfi et al. 2012). Even senescence can be induced in these cells by exposing these cells to a HDAC inhibitor such as Trichostatin A. The exact mechanisms of modulation of aging by Sirt genes are unclear, but it is hypothesized that they recruit specific chromatin remodeling factors and promote genome stability. There is also decrease in HDAC-1 expression during serial passaging of human fibroblast cells.
8.6 Change in Other Histone Modifications
Though, not very well studied, histone modifications other than DNA methylation, histone methylation and histone acetylation also have contributory effects in modulating aging like phenomenon. For example histone H2A, H2B and H4 can be modified with an O-N-acetyl-glucosamine (OGNAc). These OGNAc attached histones are enriched in gene promoters important for aging and stress responses in C. elegans (Benayoun et al. 2015). Further, deletion of the OGNAc depositing gene shortens the lifespan of C. elegans; while, deletion of the gene that removes the OGNAc mark extends the life span. Age dependent changes in the oxidative modifications of histones such as carbonylation are also reported in rat liver (Sharma et al. 2006). While, the rate of carbonylation was higher for the above mentioned histones in young rat liver, the carbomylation was significantly lower in the liver histones of aged rats. Further, dietary restriction of older rats increased carbamylation rates. A detailed change in the histone modification is represented in Table 1.
8.7 Generation of Heterochromatic Foci
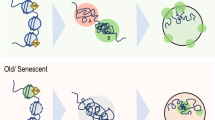
Senescent cells show discrete Senescence associated heterochromatic foci (SAHF) (Kosar et al. 2011). SAHF structures are readily stained by DAPI and show resistance to be digested by nucleases, suggesting a more compact structure. SAHF structures were first identified in aged human fibroblast cells (Glauche et al. 2011). SAHF contain chromatin domains that are resonant of the constitutive heterochromatin domains, such as enrichment of H3K9me3 and HP1and hypoacetylated histones and incorporation of macroH2A. However, these foci lack linker histone H1. It is further seen that each chromosome condenses into a single SAHF. Preliminary experiments have shown that disruption of the H3K9 methyl transferase promotes tumour formation in the lymphocytes of mice. In these cells there is prevention of senescence suggesting that SAHF complex formation is crucial for occurrence of senescence (Kosar et al. 2011).
8.8 Age Dependent Change in Chromatin Remodeling Factors
Age dependent changes in the expression of remodeling complexes are also observed, in a number of organisms. The components of NuRD (nucleosome remodeling and deacetylase) complex are found to be down regulated in aged healthy individuals as well as in HGPS patients as studied in fibroblasts (Pegoraro et al. 2009). Further, it is also demonstrated that in Hela cells, RNAi mediated down-regulation of NuRD results in loss of heterochromatin (Pegoraro et al. 2009). Similarly, in yeast and C. elegans, deletion of ISWI (chromatin remodeling imitation switch) complex increases life span (Benayoun et al. 2015). However, a functionally active SWI/SNF is required for promoting longevity in C. elegans. Few other observations also state that in human adrenal cortex carcinoma derived cell lines, senescence can be induced by BRG1. It is further seen that BrmI level increases in old mice liver correlating to age dependent repression complex formation. Further, when nuclear extract of young animals were incubated with BrmI, it resulted in the formation of similar complexes observed in aged animals (Benayoun et al. 2015).
8.9 Age Dependent Changes in Histone Variants and Histone Exchange
In non-replicating cells such as neurons, there is replication independent enrichment of histone H3.3, a histone H3 variant of the chromatin (Pina and Suau 1987). It has been further shown in chicken and mice model that during aging there is increase in H3.3 level (Urban and Zweidler 1983). This observation also correlates well with the hypothesis that aged cells have more open chromatin and hyperacetylated chromatin. In neurons, there is formation of an open chromatin due to incorporation of H3.3. In vitro also there is increased incorporation of H3.3 in human fibroblast cell lines entering aging. The H3.3 specific chaperons also increase in aged baboons (Jeyapalan et al. 2007). The histone H2A variant H2A.Z is also correlated with aging. Knockdown of H2A.Z variant or p400 (the H2A.Z exchanger) promotes aging in human fibroblasts (Chan et al. 2005).
8.10 Age Dependent Proteolysis of Histones
Two different forms of histone H3, namely a ‘slower migrating’ (H3S) and a ‘faster migrating’ (H3F) have been detected in Tetrahymena thermophila micronuclei. Using partial proteolytic peptide mapping it was suggested that the H3F was a proteolytically clipped product of H3S. It was cleaved by six amino acids residues from its N-terminal end, and it was a physiologically regulated proteolytic processing event (Allis et al. 1984). It has been further shown that when macronuclei became senescent and transcription was inactivated, the N-terminal tails of the core histones were proteolytically removed (Lin et al. 1991). It has been further proposed that the macronuclei, which were transcriptionally active, did not own such a proteolytic event.
Few recent observations have demonstrated the proteolytic processing of histone H3 and histone H1 in old rat liver and chicken (Chaturvedi, M.M. group, unpublished observation) and in Japanese quail (Mahendra et al. 1999; Mahendra and Kanungo 2000; Mishra and Kanungo 1994). It has been further shown that progesterone induces the H3 cleavage in Japanese quail, suggesting that the protease specific to histone H3 may be regulated by progesterone. Shanti, 1995, observed an additional band in the histones prepared either from nuclei or purified nucleosome-core from liver of old rats (Chaturvedi, M.M. group, unpublished observation). This band migrated between histone H2A and H4. The appearance of this band had a strong correlation with decrease in the stoichiometry of the histone H3. It was speculated that this band might be a clipped product of H3, and hence named as ΔH3. However, the precursor-product relationship between H3 and ΔH3 was not established. The ΔH3 generation has also been demonstrated recently in case of old chicken liver (Purohit et al. 2012). Through N-terminal sequencing, it has been shown that the ΔH3 was an N-terminally clipped product of histone H3, being cleaved at 23 amino acids from the N-terminus. The other histone posttranslational modification profile of the ΔH3 is presently being investigated by western blotting (Chaturvedi, M.M. group).
9 Conclusion and Future Prospects: Attaining Epigenetic Rejuvenation?
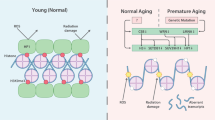
A number of increasing lines of evidence propose chromatin structure as a modulator of aging. There is aging dependent change in regulation of transcription and nuclear architecture. When organisms age, there is decline in maintenance of the cellular structure and function which brings about change in the chromatin structure and organization. However, the age dependent change in the chromatin structure is multifaceted and often antagonistic in different organisms. For instance, in most of the organisms, with aging there is decreased histone expression and decrease in heterochromatin abundance with concomitant formation of an open chromatin structure. However, in the replicating senescence model there is age dependent accumulation of heterochromatic foci. The other examples also include the redistribution of SIR proteins in yeast during aging from the telomere and MAT locus to the rDNA regions promoting rDNA silencing and telomere shortening. At present restoring back to a youthful chromatin structure involves elevation in histone expression, reduction in acetylation of H3-N-terminal and H4K16 and inactivation of the HDAC Rpd3.
The epigenome is also influenced by environmental factors. Throughout the lifespan of an organism, the insults induced by the environment also contribute to the functioning of cells to organs. Thus it can also be proposed that the epigenomic instability induced by environment and life style also influence aging as represented by the methylation clock. It can be further stated that modulation of the epigenetic reprogramming by altering chromatin remodeling factors can revert aging. Hence, these chromatin remodeling factors pose a good candidate for therapeutic interventions. A detailed account of the aging mediated changes is summarised in Fig. 2.

Summary of the changes in the chromatin organization and function in aged cells. Normal cells have distinct perinuclear heterochromatin foci and few facultative heterochromatin loci. Aged cells on the contrary have disorganized perinuclear heterochromatin with concomitant increase in facultative heterochromatin loci. Also, in contrast to the Lamin A deposition in the nuclear membrane and interaction of NuRD complex to lamin A in young cells, there is an age dependent accumulation of progerin leading to disorganization of the nuclear architecture. In addition, there is decreased expression of NuRD complex in aged cells. Further, in aged cells, there is decreased core histone expression leading to reduced histone occupancy in the nucleosomal array. In addition to that there are a number of changes in the histone and DNA PTM, such as methylation and acetylation in the promoter and the coding region of the genes, which are indicated
However, several questions related to aging are still unclear and require future investigation. 1. The change in chromatin modifications are the cause or the consequence of aging? 2. Could the epigenetic players be precisely modulated and could act as therapeutic targets for reverting aging? 3. Since, the epigenetic modulators are broadly global; could they be specifically managed by independent linear pathway so that when therapeutic interventions are formulated, they will have minimal side effects. We are sure, answers to these questions are crucial before formulation of any medicine/agents for epigenetic rejuvenation. Few recent reports represent that HP1β and mH2A are the prime targets in this respect. However, the concept of epigenetic rejuvenation is still in its infancy and needs manifold unravelling in near future.
References
Adams PD (2007) Remodeling of chromatin structure in senescent cells and its potential impact on tumor suppression and aging. Gene 397:84–93
Allis CD, Allen RL, Wiggins JC, Chicoine LG, Richman R (1984) Proteolytic processing of h1-like histones in chromatin: a physiologically and developmentally regulated event in Tetrahymena micronuclei. J Cell Biol 99:1669–1677
Allis CD, Bowen JK, Abraham GN, Glover CV, Gorovsky MA (1980) Proteolytic processing of histone H3 in chromatin: a physiologically regulated event in Tetrahymena micronuclei. Cell 20:55–64
Ashok BT, Ali R (2003) Aging research in India. Exp Gerontol 38:597–603
Bannister AJ, Kouzarides T (2015) Regulation of chromatin by histone modifications. Cell Res 21:381–395
Bannister AJ, Schneider R, Kouzarides T (2002) Histone methylation: dynamic or static? Cell 109:801–806
Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410:120–124
Bao Y, Shen X (2007) INO80 subfamily of chromatin remodeling complexes. Mutat Res 618:18–29
Baumgart M et al (2015) RNA-seq of the aging brain in the short-lived fish N. furzeri—conserved pathways and novel genes associated with neurogenesis. Aging Cell 13:965–974
Bedford MT, Richard S (2005) Arginine methylation an emerging regulator of protein function. Mol Cell 18:263–272
Bednar J, Horowitz RA, Grigoryev SA, Carruthers LM, Hansen JC, Koster AJ, Woodcock CL (1998) Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc Natl Acad Sci USA 95:14173–14178
Belz T, Pham AD, Beisel C, Anders N, Bogin J, Kwozynski S, Sauer F (2002) In vitro assays to study protein ubiquitination in transcription. Methods 26:233–244
Benayoun BA, Pollina EA, Brunet A (2015) Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol 16:593–610
Berger SL (2002) Histone modifications in transcriptional regulation. Curr Opin Genet Dev 12:142–148
Bernstein BE et al (2005) Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120:169–181
Bi X, Broach JR (2001) Chromosomal boundaries in S. cerevisiae. Curr Opin Genet Dev 11:199–204
Brandt WF, Bohm L, Von Holt C (1975) Proteolytic degradation of histones and site of cleavage in histone F2al and F3. FEBS Lett 51:88–93
Briggs SD, Bryk M, Strahl BD, Cheung WL, Davie JK, Dent SY, Winston F, Allis CD (2001) Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev 15:3286–3295
Cairns BR, Lorch Y, Li Y, Zhang M, Lacomis L, Erdjument-Bromage H, Tempst P, Du J, Laurent B, Kornberg RD (1996) RSC, an essential, abundant chromatin-remodeling complex. Cell 87:1249–1260
Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y (2002) Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298:1039–1043
Carrozza MJ, Florens L, Swanson SK, Shia WJ, Anderson S, Yates J, Washburn MP, Workman JL (2005) Stable incorporation of sequence specific repressors Ash1 and Ume6 into the Rpd3L complex. Biochim Biophys Acta 1731:77–87; discussion 75–76
Carruthers LM, Bednar J, Woodcock CL, Hansen JC (1998) Linker histones stabilize the intrinsic salt-dependent folding of nucleosomal arrays: mechanistic ramifications for higher-order chromatin folding. Biochemistry 37:14776–14787
Cattanach BM (1974) Position effect variegation in the mouse. Genet Res 23:291–306
Chan HM, Narita M, Lowe SW, Livingston DM (2005) The p400 E1A-associated protein is a novel component of the p53 --> p21 senescence pathway. Genes Dev 19:196–201
Chang B, Chen Y, Zhao Y, Bruick RK (2007) JMJD6 is a histone arginine demethylase. Science 318:444–447
Chaturvedi MM, Kanungo MS (1985) Analysis of conformation and function of the chromatin of the brain of young and old rats. Mol Biol Rep 10:215–219
Clapier CR, Cairns BR (2009) The biology of chromatin remodeling complexes. Annu Rev Biochem 78:273–304
Conaway RC, Brower CS, Conaway JW (2002) Emerging roles of ubiquitin in transcription regulation. Science 296:1254–1258
Cuthbert GL et al (2004) Histone deimination antagonizes arginine methylation. Cell 118:545–553
Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V (2002) Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111:185–196
Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, Berger SL (2009) Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459:802–807
Daser A, Rabbitts TH (2004) Extending the repertoire of the mixed-lineage leukemia gene MLL in leukemogenesis. Genes Dev 18:965–974
de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB (2003) Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 370:737–749
Dechassa ML, Sabri A, Pondugula S, Kassabov SR, Chatterjee N, Kladde MP, Bartholomew B (2010) SWI/SNF has intrinsic nucleosome disassembly activity that is dependent on adjacent nucleosomes. Mol Cell 38:590–602
DeLange RJ, Fambrough DM, Smith EL, Bonner J (1969) Calf and pea histone IV. II. The complete amino acid sequence of calf thymus histone IV; presence of epsilon-N-acetyllysine. J Biol Chem 244:319–334
Delmas V, Stokes DG, Perry RP (1993) A mammalian DNA-binding protein that contains a chromodomain and an SNF2/SWI2-like helicase domain. Proc Natl Acad Sci USA 90:2414–2418
Dhaenens M, Glibert P, Meert P, Vossaert L, Deforce D (2015) Histone proteolysis: a proposal for categorization into ‘clipping’ and ‘degradation’. BioEssays 37:70–79
Dirscherl SS, Krebs JE (2004) Functional diversity of ISWI complexes. Biochem Cell Biol 82:482–489
Duncan EM, Muratore-Schroeder TL, Cook RG, Garcia BA, Shabanowitz J, Hunt DF, Allis CD (2008) Cathepsin L proteolytically processes histone H3 during mouse embryonic stem cell differentiation. Cell 135:284–294
Eickbush TH, Godfrey JE, Elia MC, Moudrianakis EN (1988) H2a-specific proteolysis as a unique probe in the analysis of the histone octamer. J Biol Chem 263:18972–18978
Eisenberg T et al (2009) Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 11:1305–1314
Etzioni R, Legler JM, Feuer EJ, Merrill RM, Cronin KA, Hankey BF (1999) Cancer surveillance series: interpreting trends in prostate cancer–part III: Quantifying the link between population prostate-specific antigen testing and recent declines in prostate cancer mortality. J Natl Cancer Inst 91:1033–1039
Falk MM, Grigera PR, Bergmann IE, Zibert A, Multhaup G, Beck E (1990) Foot-and-mouth disease virus protease 3C induces specific proteolytic cleavage of host cell histone H3. J Virol 64:748–756
Faulkner RD, Bhatnagar YM (1987) A protease activity is associated with testicular chromatin of the mouse. Biol Reprod 36:471–480
Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, Zhang Y (2002) Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol 12:1052–1058
Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, Tyler JK (2010) Elevated histone expression promotes life span extension. Mol Cell 39:724–735
Feser J, Tyler J (2011) Chromatin structure as a mediator of aging. FEBS Lett 585:2041–2048
Fletcher TM, Hansen JC (1996) The nucleosomal array: structure/function relationships. Crit Rev Eukaryot Gene Expr 6:149–188
Fugita OE, Chan DY, Roberts WW, Kavoussi LR, Jarrett TW (2004). Laparoscopic radical nephrectomy in obese patients: outcomes and technical considerations. Urology 63:247–252; discussion 252
Gary JD, Clarke S (1998) RNA and protein interactions modulated by protein arginine methylation. Prog Nucleic Acid Res Mol Biol 61:65–131
Gaubatz JW, Cutler RG (1990) Mouse satellite DNA is transcribed in senescent cardiac muscle. J Biol Chem 265:17753–17758
Gerber M, Shilatifard A (2003) Transcriptional elongation by RNA polymerase II and histone methylation. J Biol Chem 278:26303–26306
Gershey EL, Haslett GW, Vidali G, Allfrey VG (1969) Chemical studies of histone methylation. Evidence for the occurrence of 3-methylhistidine in avian erythrocyte histone fractions. J Biol Chem 244:4871–4877
Girdwood D, Bumpass D, Vaughan OA, Thain A, Anderson LA, Snowden AW, Garcia-Wilson E, Perkins ND, Hay RT (2003) P300 transcriptional repression is mediated by SUMO modification. Mol Cell 11:1043–1054
Glauche I, Thielecke L, Roeder I (2011) Cellular aging leads to functional heterogeneity of hematopoietic stem cells: a modeling perspective. Aging Cell 10:457–465
Gravina S, Vijg J (2010) Epigenetic factors in aging and longevity. Pflugers Arch 459:247–258
Greenwell PW, Kronmal SL, Porter SE, Gassenhuber J, Obermaier B, Petes TD (1995) TEL1, a gene involved in controlling telomere length in S. cerevisiae, is homologous to the human ataxia telangiectasia gene. Cell 82:823–829
Greer EL, Maures TJ, Ucar D, Hauswirth AG, Mancini E, Lim JP, Benayoun BA, Shi Y, Brunet A (2011) Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature 479:365–371
Grigera PR, Tisminetzky SG (1984) Histone H3 modification in BHK cells infected with foot-and-mouth disease virus. Virology 136:10–19
Grigoryev SA (2001) Higher-order folding of heterochromatin: protein bridges span the nucleosome arrays. Biochem Cell Biol 79:227–241
Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL (2001) Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J Biol Chem 276:38837–38843
Hakimi MA, Bochar DA, Chenoweth J, Lane WS, Mandel G, Shiekhattar R (2002) A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc Natl Acad Sci USA 99:7420–7425
Hampsey M, Reinberg D (2003) Tails of intrigue: phosphorylation of RNA polymerase II mediates histone methylation. Cell 113:429–432
Hanks SK, Rodriguez LV, Rao PN (1983) Relationship between histone phosphorylation and premature chromosome condensation. Exp Cell Res 148:293–302
Hansen JC (2002) Conformational dynamics of the chromatin fiber in solution: determinants, mechanisms, and functions. Annu Rev Biophys Biomol Struct 31:361–392
Hirano T (2000) Chromosome cohesion, condensation, and separation. Annu Rev Biochem 69:115–144
Hong L, Schroth GP, Matthews HR, Yau P, Bradbury EM (1993) Studies of the DNA binding properties of histone H4 amino terminus. Thermal denaturation studies reveal that acetylation markedly reduces the binding constant of the H4 “tail” to DNA. J Biol Chem 268:305–314
Hu Z, Chen K, Xia Z, Chavez M, Pal S, Seol JH, Chen CC, Li W, Tyler JK (2014) Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev 28:396–408
Imai S, Armstrong CM, Kaeberlein M, Guarente L (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403:795–800
Jackson JP, Lindroth AM, Cao X, Jacobsen SE (2002) Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature 416:556–560
Jacobson MK, Jacobson EL (1999) Discovering new ADP-ribose polymer cycles: protecting the genome and more. Trends Biochem Sci 24:415–417
Jentsch S, Schlenker S (1995) Selective protein degradation: a journey’s end within the proteasome. Cell 82:881–884
Jenuwein T, Allis CD (2001) Translating the histone code. Science 293:1074–1080
Jeyapalan JC, Ferreira M, Sedivy JM, Herbig U (2007) Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev 128:36–44
Jones PA, Baylin SB (2002) The fundamental role of epigenetic events in cancer. Nat Rev Genet 3:415–428
Jurkowski TP, Jeltsch A (2011) Burning off DNA methylation: new evidence for oxygen-dependent DNA demethylation. ChemBioChem 12:2543–2545
Kaeberlein M, McVey M, Guarente L (1999) The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13:2570–2580
Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, Cohen HY (2012) The sirtuin SIRT6 regulates lifespan in male mice. Nature 483:218–221
Kappus S, Apweiler R, White CJ, Whish WJ (1993) In vitro poly-(ADP-ribosyl)ation of chromatin proteins in the rat tapeworm, Hymenolepis diminuta. Comp Biochem Physiol B 104:711–716
Kennedy BK, Gotta M, Sinclair DA, Mills K, McNabb DS, Murthy M, Pak SM, Laroche T, Gasser SM, Guarente L (1997) Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell 89:381–391
Kornberg RD (1974) Chromatin structure: a repeating unit of histones and DNA. Science 184:868–871
Kornberg RD, Thomas JO (1974) Chromatin structure; oligomers of the histones. Science 184:865–868
Kosar M, Bartkova J, Hubackova S, Hodny Z, Lukas J, Bartek J (2011) Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16(ink4a). Cell Cycle 10:457–468
Kouzarides T (2002) Histone methylation in transcriptional control. Curr Opin Genet Dev 12:198–209
Krishnan V, Chow MZ, Wang Z, Zhang L, Liu B, Liu X, Zhou Z (2011) Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc Natl Acad Sci USA 108:12325–12330
Krogan NJ, Dover J, Khorrami S, Greenblatt JF, Schneider J, Johnston M, Shilatifard A (2002) COMPASS, a histone H3 (Lysine 4) methyltransferase required for telomeric silencing of gene expression. J Biol Chem 277:10753–10755
Krogan NJ et al (2003) Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol Cell Biol 23:4207–4218
Kuo MH, Brownell JE, Sobel RE, Ranalli TA, Cook RG, Edmondson DG, Roth SY, Allis CD (1996) Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature 383:269–272
Lachner M, O’Sullivan RJ, Jenuwein T (2003) An epigenetic road map for histone lysine methylation. J Cell Sci 116:2117–2124
Lacoste N, Utley RT, Hunter JM, Poirier GG, Cote J (2002) Disruptor of telomeric silencing-1 is a chromatin-specific histone H3 methyltransferase. J Biol Chem 277:30421–30424
Larson K, Yan SJ, Tsurumi A, Liu J, Zhou J, Gaur K, Guo D, Eickbush TH, Li WX (2012) Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLoS Genet 8:e1002473
Le Rhun Y, Kirkland JB, Shah GM (1998) Cellular responses to DNA damage in the absence of Poly(ADP-ribose) polymerase. Biochem Biophys Res Commun 245:1–10
Lee DY, Hayes JJ, Pruss D, Wolffe AP (1993) A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 72:73–84
Lehnertz B, Ueda Y, Derijck AA, Braunschweig U, Perez-Burgos L, Kubicek S, Chen T, Li E, Jenuwein T, Peters AH (2003) Suv39 h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 13:1192–1200
Lesur I, Campbell JL (2004) The transcriptome of prematurely aging yeast cells is similar to that of telomerase-deficient cells. Mol Biol Cell 15:1297–1312
Li G, Reinberg D (2011) Chromatin higher-order structures and gene regulation. Curr Opin Genet Dev 21:175–186
Li X et al (2014) Histone demethylase KDM5B is a key regulator of genome stability. Proc Natl Acad Sci USA 111:7096–7101
Lin R, Cook RG, Allis CD (1991) Proteolytic removal of core histone amino termini and dephosphorylation of histone H1 correlate with the formation of condensed chromatin and transcriptional silencing during Tetrahymena macronuclear development. Genes Dev 5:1601–1610
Liu L, Cheung TH, Charville GW, Hurgo BM, Leavitt T, Shih J, Brunet A, Rando TA (2013) Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep 4:189–204
Luger K, Dechassa ML, Tremethick DJ (2012) New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat Rev Mol Cell Biol 13:436–447
Luger K, Rechsteiner TJ, Flaus AJ, Waye MM, Richmond TJ (1997) Characterization of nucleosome core particles containing histone proteins made in bacteria. J Mol Biol 272:301–311
Macieira-Coelho A, Puvion-Dutilleul F (1989) Evaluation of the reorganization in the high-order structure of DNA occurring during cell senescence. Mutat Res 219:165–170
Mahendra G, Gupta S, Kanungo MS (1999) Effect of 17beta estradiol and progesterone on the conformation of the chromatin of the liver of female Japanese quail during aging. Arch Gerontol Geriatr 28:149–158
Mahendra G, Kanungo MS (2000) Age-related and steroid induced changes in the histones of the quail liver. Arch Gerontol Geriatr 30:109–114
Marchal C, Miotto B (2015) Emerging concept in DNA methylation: role of transcription factors in shaping DNA methylation patterns. J Cell Physiol 230:743–751
Marushige K, Marushige Y, Wong TK (1976) Complete displacement of somatic histones during transformation of spermatid chromatin: a model experiment. Biochemistry 15:2047–2053
Marushige Y, Marushige K (1983) Proteolysis of somatic type histones in transforming rat spermatid chromatin. Biochim Biophys Acta 761:48–57
Maures TJ, Greer EL, Hauswirth AG, Brunet A (2011) The H3K27 demethylase UTX-1 regulates C. elegans lifespan in a germline-independent, insulin-dependent manner. Aging Cell 10:980–990
McColl G, Killilea DW, Hubbard AE, Vantipalli MC, Melov S, Lithgow GJ (2008) Pharmacogenetic analysis of lithium-induced delayed aging in Caenorhabditis elegans. J Biol Chem 283:350–357
Melchior F (2000) SUMO–nonclassical ubiquitin. Annu Rev Cell Dev Biol 16:591–626
Meshorer E, Gruenbaum Y (2009) NURD keeps chromatin young. Nat Cell Biol 11:1176–1177
Metzger E, Wissmann M, Schule R (2006) Histone demethylation and androgen-dependent transcription. Curr Opin Genet Dev 16:513–517
Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A (2001) COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci USA 98:12902–12907
Mishra RN, Kanungo MS (1994) Alterations in histones of the liver and oviduct of Japanese quail during aging. Mol Biol Rep 20:15–18
Morrison AJ et al (2007) Mec1/Tel1 phosphorylation of the INO80 chromatin remodeling complex influences DNA damage checkpoint responses. Cell 130:499–511
Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O’Connor MB, Kingston RE, Simon JA (2002) Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111:197–208
Murray K (1964) The Occurrence of Epsilon-N-Methyl Lysine in Histones. Biochemistry 3:10–15
Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI (2001) Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292:110–113
Nathan D, Sterner DE, Berger SL (2003) Histone modifications: Now summoning sumoylation. Proc Natl Acad Sci USA 100:13118–13120
Ng HH, Feng Q, Wang H, Erdjument-Bromage H, Tempst P, Zhang Y, Struhl K (2002) Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev 16:1518–1527
Ni Z, Ebata A, Alipanahiramandi E, Lee SS (2012) Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans. Aging Cell 11:315–325
Nielsen PR, Nietlispach D, Mott HR, Callaghan J, Bannister A, Kouzarides T, Murzin AG, Murzina NV, Laue ED (2002) Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature 416:103–107
O’Carroll D et al (2000) Isolation and characterization of Suv39h2, a second histone H3 methyltransferase gene that displays testis-specific expression. Mol Cell Biol 20:9423–9433
Oberdoerffer P, Sinclair DA (2007) The role of nuclear architecture in genomic instability and ageing. Nat Rev Mol Cell Biol 8:692–702
Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87:953–959
Okawa Y, Takada K, Minami J, Aoki K, Shibayama H, Ohkawa K (2003) Purification of N-terminally truncated histone H2A-monoubiquitin conjugates from leukemic cell nuclei: probable proteolytic products of ubiquitinated H2A. Int J Biochem Cell Biol 35:1588–1600
Olins DE, Wright EB (1973) Glutaraldehyde fixation of isolated eucaryotic nuclei. Evidence for histone-histone proximity. J Cell Biol 59:304–317
Padilla PA, Garcia AM, Ladage ML, Toni LS (2014) Caenorhabditis elegans: an old genetic model can learn new epigenetic tricks. Integr Comp Biol 54:52–60
Panda P, Chaturvedi MM, Panda AK, Suar M, Purohit JS (2013) Purification and characterization of a novel histone H2A specific protease (H2Asp) from chicken liver nuclear extract. Gene 512:47–54
Panigrahi AK, Tomar RS, Chaturvedi MM (2003) A SWI/SNF-like factor from chicken liver that disrupts nucleosomes and transfers histone octamers in cis and trans. Arch Biochem Biophys 414:24–33
Patterson BD, Davies DD (1969) Specificity of the enzymatic methylation of pea histone. Biochem Biophys Res Commun 34:791–794
Pegoraro G, Kubben N, Wickert U, Gohler H, Hoffmann K, Misteli T (2009) Ageing-related chromatin defects through loss of the NURD complex. Nat Cell Biol 11:1261–1267
Peters AH et al (2003) Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell 12:1577–1589
Peters AH et al (2001) Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107:323–337
Pfaffeneder T, Hackner B, Truss M, Munzel M, Muller M, Deiml CA, Hagemeier C, Carell T (2011) The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew Chem Int Ed Engl 50:7008–7012
Pina B, Suau P (1987) Changes in histones H2A and H3 variant composition in differentiating and mature rat brain cortical neurons. Dev Biol 123:51–58
Pray-Grant MG, Daniel JA, Schieltz D, Yates JR 3rd, Grant PA (2005) Chd1 chromodomain links histone H3 methylation with SAGA- and SLIK-dependent acetylation. Nature 433:434–438
Pu M, Ni Z, Wang M, Wang X, Wood JG, Helfand SL, Yu H, Lee SS (2015) Trimethylation of Lys36 on H3 restricts gene expression change during aging and impacts life span. Genes Dev 29:718–731
Purohit JS, Chaturvedi MM, Panda P (2012) Histone proteases: the tale of tail clippers. Int J Intgv Sci Innov Tech 1:51–60
Purohit JS, Tomar RS, Panigrahi AK, Pandey SM, Singh D, Chaturvedi MM (2013) Chicken liver glutamate dehydrogenase (GDH) demonstrates a histone H3 specific protease (H3ase) activity in vitro. Biochimie 95:1999–2009
Rea S et al (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406:593–599
Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, Shinkai Y, Allis CD (2003) Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell 12:1591–1598
Ringrose L, Paro R (2004) Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet 38:413–443
Robinson PJ, Fairall L, Huynh VA, Rhodes D (2006) EM measurements define the dimensions of the “30-nm” chromatin fiber: evidence for a compact, interdigitated structure. Proc Natl Acad Sci U S A 103:6506–6511
Robzyk K, Recht J, Osley MA (2000) Rad6-dependent ubiquitination of histone H2B in yeast. Science 287:501–504
Roguev A, Schaft D, Shevchenko A, Pijnappel WW, Wilm M, Aasland R, Stewart AF (2001) The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J 20:7137–7148
Roth SY, Denu JM, Allis CD (2001) Histone acetyltransferases. Annu Rev Biochem 70:81–120
Sarraf SA, Stancheva I (2004) Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol Cell 15:595–605
Scaffidi P, Misteli T (2006) Lamin A-dependent nuclear defects in human aging. Science 312:1059–1063
Schneider J, Wood A, Lee JS, Schuster R, Dueker J, Maguire C, Swanson SK, Florens L, Washburn MP, Shilatifard A (2005) Molecular regulation of histone H3 trimethylation by COMPASS and the regulation of gene expression. Mol Cell 19:849–856
Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T (2004) A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev 18:1251–1262
Sedivy JM, Banumathy G, Adams PD (2008) Aging by epigenetics–a consequence of chromatin damage? Exp Cell Res 314:1909–1917
Sen P et al (2015) H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev 29:1362–1376
Shah PP et al (2013) Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev 27:1787–1799
Sharma R, Nakamura A, Takahashi R, Nakamoto H, Goto S (2006) Carbonyl modification in rat liver histones: decrease with age and increase by dietary restriction. Free Radic Biol Med 40:1179–1184
Shenker N, Flanagan JM (2011) Intragenic DNA methylation: implications of this epigenetic mechanism for cancer research. Br J Cancer 106:248–253
Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119:941–953
Shi Y, Sawada J, Sui G, el Affar B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y (2003) Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422:735–738
Shiio Y, Eisenman RN (2003) Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci U S A 100:13225–13230
Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL (2006) Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311:844–847
Singer MS, Kahana A, Wolf AJ, Meisinger LL, Peterson SE, Goggin C, Mahowald M, Gottschling DE (1998) Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics 150:613–632
Smeal T, Claus J, Kennedy B, Cole F, Guarente L (1996) Loss of transcriptional silencing causes sterility in old mother cells of S. cerevisiae. Cell 84:633–642
Smith ST, Petruk S, Sedkov Y, Cho E, Tillib S, Canaani E, Mazo A (2004) Modulation of heat shock gene expression by the TAC1 chromatin-modifying complex. Nat Cell Biol 6:162–167
Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403:41–45
Strahl BD et al (2002) Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol Cell Biol 22:1298–1306
Straight AF, Shou W, Dowd GJ, Turck CW, Deshaies RJ, Johnson AD, Moazed D (1999) Net1, a Sir2-associated nucleolar protein required for rDNA silencing and nucleolar integrity. Cell 97:245–256
Struhl K (1998) Histone acetylation and transcriptional regulatory mechanisms. Genes Dev 12:599–606
Suka N, Suka Y, Carmen AA, Wu J, Grunstein M (2001) Highly specific antibodies determine histone acetylation site usage in yeast heterochromatin and euchromatin. Mol Cell 8:473–479
Sun ZW, Allis CD (2002) Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 418:104–108
Tachibana M, Sugimoto K, Fukushima T, Shinkai Y (2001) Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J Biol Chem 276:25309–25317
Tamaru H, Selker EU (2001) A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature 414:277–283
Tanaka Y, Fujii T, Yamana H, Kato S, Morimatsu M, Shirouzu K (2004) Experimental gene therapy using p21Waf1 gene for esophageal squamous cell carcinoma by gene gun technology. Int J Mol Med 14:545–551
Tesar M, Marquardt O (1990) Foot-and-mouth disease virus protease 3C inhibits cellular transcription and mediates cleavage of histone H3. Virology 174:364–374
Timmermann S, Lehrmann H, Polesskaya A, Harel-Bellan A (2001) Histone acetylation and disease. Cell Mol Life Sci 58:728–736
Timmers HT, Tora L (2005) SAGA unveiled. Trends Biochem Sci 30:7–10
Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y (2006) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439:811–816
Tsurumi A, Li WX (2012) Global heterochromatin loss: a unifying theory of aging? Epigenetics 7:680–688
Turner BM (2000) Histone acetylation and an epigenetic code. BioEssays 22:836–845
Urban MK, Zweidler A (1983) Changes in nucleosomal core histone variants during chicken development and maturation. Dev Biol 95:421–428
Vakoc CR, Mandat SA, Olenchock BA, Blobel GA (2005) Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol Cell 19:381–391
van Holde K, Zlatanova J (1995) Chromatin higher order structure: chasing a mirage? J Biol Chem 270:8373–8376
van Leeuwen F, Gafken PR, Gottschling DE (2002) Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 109:745–756
Vaquero A, Loyola A, Reinberg D (2003) The constantly changing face of chromatin. Sci Aging Knowl Environ 2003 RE4
Vignali M, Hassan AH, Neely KE, Workman JL (2000) ATP-dependent chromatin-remodeling complexes. Mol Cell Biol 20:1899–1910
Vire E et al (2006) The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439:871–874
Vogelauer M, Wu J, Suka N, Grunstein M (2000) Global histone acetylation and deacetylation in yeast. Nature 408:495–498
Whetstine JR et al (2006) Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125:467–481
Wood JG, Helfand SL (2013) Chromatin structure and transposable elements in organismal aging. Front Genet 4:274
Woodage T, Basrai MA, Baxevanis AD, Hieter P, Collins FS (1997) Characterization of the CHD family of proteins. Proc Natl Acad Sci USA 94:11472–11477
Woodcock CL, Ghosh RP (2010) Chromatin Higher-order Structure and Dynamics. Cold Spring Harbor Perspectives in Biology 2
Woodcock CL, Grigoryev SA, Horowitz RA, Whitaker N (1993) A chromatin folding model that incorporates linker variability generates fibers resembling the native structures. Proc Natl Acad Sci USA 90:9021–9025
Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, Roeder RG, Brivanlou AH, Allis CD (2005) WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 121:859–872
Xu W, Edmondson DG, Roth SY (1998) Mammalian GCN5 and P/CAF acetyltransferases have homologous amino-terminal domains important for recognition of nucleosomal substrates. Mol Cell Biol 18:5659–5669
Xu Y et al (2011) Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol Cell 42:451–464
Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W (1998) NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell 2:851–861
Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y (2006) JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 125:483–495
Yang L, Xia L, Wu DY, Wang H, Chansky HA, Schubach WH, Hickstein DD, Zhang Y (2002) Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene 21:148–152
Yang SY, Yang XL, Yao LF, Wang HB, Sun CK (2013) Effect of CpG methylation on DNA binding protein: molecular dynamics simulations of the homeodomain PITX2 bound to the methylated DNA. J Mol Graph Model 29:920–927
Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y (1996) A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature 382:319–324
You A, Tong JK, Grozinger CM, Schreiber SL (2001) CoREST is an integral component of the CoREST—human histone deacetylase complex. Proc Natl Acad Sci U S A 98:1454–1458
Zeng L, Zhou MM (2002) Bromodomain: an acetyl-lysine binding domain. FEBS Lett 513:124–128
Zhang Y, Reinberg D (2001) Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev 15:2343–2360
Zou J, Lippert C, Heckerman D, Aryee M, Listgarten J (2014) Epigenome-wide association studies without the need for cell-type composition. Nat Methods 11:309–311
Acknowledgments
MMC and JSP acknowledge the financial support extended to them by Delhi University, form DST purse grant and R&D grants of Delhi University for their research.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media Singapore
About this chapter
Cite this chapter
Purohit, J.S., Chaturvedi, M.M. (2017). Chromatin and Aging. In: Rath, P., Sharma, R., Prasad, S. (eds) Topics in Biomedical Gerontology. Springer, Singapore. https://doi.org/10.1007/978-981-10-2155-8_11
Download citation
DOI: https://doi.org/10.1007/978-981-10-2155-8_11
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-2154-1
Online ISBN: 978-981-10-2155-8
eBook Packages: MedicineMedicine (R0)