
Abstract
Over the past two decades, cell-penetrating peptides (CPPs) have become increasingly popular both in research and in application. There have been numerous studies on the physiochemical characteristics and behavior of CPPs in various environments; likewise, the mechanisms of entry and delivery capabilities of these peptides have also been extensively researched. Besides the fundamental issues, there is an enormous interest in the delivery capabilities of the peptides as the family of CPPs is a promising and mostly non-toxic delivery vector candidate for numerous medical applications such as gene silencing, transgene delivery, and splice correction. Lately, however, there has been an emerging field of study besides the high-profile gene therapy applications—the use of peptides and CPPs to combat various infections caused by harmful bacteria, fungi, and viruses.
In this chapter, we aim to provide a short overview of the history and properties of CPPs which is followed by more thorough descriptions of antimicrobial and antiviral peptides. To achieve this, we analyze the origin of such peptides, give an overview of the mechanisms of action and discuss the various practical applications which are ongoing or have been suggested based on research.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others
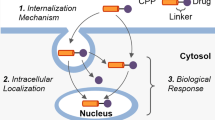

Key words
1 Introduction
CPPs are a class of diverse peptides, typically 5–30 amino acids in length, and unlike most peptides, they can cross the cellular plasma membrane. CPPs are a family of peptides that are structurally diverse but share the ability to translocate a wide range of different bioactive molecules into living cells [1]. The list of available CPPs has grown rapidly and CPPs have been employed for a variety of applications. CPPs serving as vectors can successfully facilitate the intracellular transport of cargoes, such as small-molecule therapeutic agents [2], proteins, quantum dots [3], and MRI contrast agents [4], both in vitro and in vivo [5]. In addition, this efficient transport system has lower cytotoxicity in a variety of cell lines compared with other delivery methods [6].
The mechanism of the internalization of CPPs and their cargo is not well understood and has recently been the subject of controversy. CPPs can interact with multiple cell surface molecules, including membrane lipids and membrane-associated proteoglycans [7]. CPPs can be taken up by cells via multiple pathways, such as direct translocation through the membrane bilayer or endocytosis-mediated uptake. Although molecules entering cells prefer the direct membrane translocation pathway, endocytosis, composed of two steps, endocytotic entry and endosomal escape, is the major cellular uptake pathway for most CPPs as most reports have pointed out [8]. Endocytosis occurs by various formats, which can be classified into caveolae and/or lipid raft-mediated endocytosis [9], micropinocytosis [10], through clathrin-mediated endocytosis [11] or via a cholesterol-dependent clathrin-mediated endocytosis [12]. The endocytosis pathway is proposed based on the fact that CPP uptake in cells was found to adopt an energy-dependent mechanism [13]. However, cellular uptake is not completely prohibited at low temperatures or in the presence of endocytosis inhibitors [13], suggesting a non-energy consuming route. Recently, the finding of strong peptide–lipid interactions supports direct translocation of the peptides across the membrane [14]. In addition, many studies also propose cellular uptake can follow multiple routes depending on experimental conditions, specific CPP sequence, and the cargo structure [15].
Besides CPPs, another important category of cationic membrane peptides are antimicrobial peptides (AMPs). Antimicrobial peptides, a major class of antibacterial agents, share amphiphilicity and cationic structural properties with cell-penetrating peptides. All AMPs known by the late-1990s are cationic. However, the concept that AMPs need to be cationic was changed later with the discovery of negatively charged AMPs in 1997 [16]. For example, maximin-H5 [17] discovered from frog skin and dermicidin [18] secreted from human sweat gland tissues are both anionic peptides. They also have a positive net charge, normally in the range of +4 to +6, which is due to the frequent presence of lysines and arginines in the amino acid sequence [19]. Additionally, nearly 50 % of their structure often consists of hydrophobic residues. They kill a broad spectrum of microbes including bacteria and fungi by destroying their cell membranes and are important components of the innate immune system of many animals and plants [20].
2 CPPs and AMPs: How Different Are They?
CPPs and AMPs share common characteristics. CPPs and AMPs are similar in the structural motif of amino acid composition, but differ in biological activities within the lipid membrane. Common to both groups are interactions of cell membrane components with the charged amino acid residues of the peptides. This is probably the first step in cell association that leads to cellular uptake. Anionic phospholipids or phosphate groups of lipopolysaccharides (for gram-negative bacteria) or acidic polysaccharides, teichoic acids, and lipoteichoic acids (for gram-positive bacteria) are the membrane components at the cell surface responsible for generating an overall negative net charge, making the binding of positively charged or amphipathic peptides possible [21]. Furthermore, the lipid bilayer of bacterial membrane contains mainly lipids with negatively charged phospholipid headgroups, while fungi, in contrast, exhibit a zwitterionic lipid bilayer composition, upon which the uptake is driven by hydrophobic N- or C-terminus or particular amino acids of the peptide sequence, resulting in the accumulation of the peptide, which finally induces strong hydrophobic interaction to the membrane.
Charge interactions between cationic residues and lipids are essential to the biological function of many membrane peptides and proteins, for example, cellular translocation of CPPs, and membrane disruption of AMPs. The ratio between the cationic lysine (Lys) and arginine (Arg) residues influences membrane selectivity since the guanidino functionalities of arginines promote a more efficient interaction with eukaryotic membranes as compared to lysine. This is, however, most often at the expense of increased cytotoxicity. A high Lys content has been correlated with selectivity toward bacterial cells over eukaryotic cells [22]. Furthermore, the ability of CPPs to permeate cell membranes appears to be directly linked to their propensity to fold into a well-defined secondary structure (α-helix or β-sheet) while interacting with biological membranes [23]. Likewise, the antimicrobial activity of α-helical AMPs depends on their propensity to form an α-helix [24]. The highly basic Arg guanidinium and Lys ammonium groups remain protonated under physiological pH conditions and thus can function as hydrogen bond (H-bond) donors in various protein–protein and protein–lipid interactions. In lipid membranes, the guanidinium group in protein side chain can form a branched moiety by interacting with water and lipid molecules. In the past few years, many CPPs and AMPs have been extensively investigated to elucidate the structural basis of how these cationic macromolecules interact with membrane lipids and water [25].
Two examples of AMPs in the recent studies include protegrin-1 (PG-1) [26] and human neutrophil peptide-1 (HNP-1) [27]. PG-1 is representative of many β-sheet AMPs in its disulfide-linked structure and has an Arg-rich sequence. HNP-1 belongs to the α-defensin family of antimicrobial peptides and is the mediator of the host innate immune response. Most AMPs adopt rigid amphipathic secondary structures, either α-helical or β-sheet [28]. The two CPPs, TAT and penetratin, are the first two discovered and also most frequently applied CPPs [25]. They display a turn-rich conformation and random coil structure in lipid bilayers, respectively, suggesting that the absence of intra- or intermolecular H-bonded conformation and high molecular mobility may be the hallmarks of CPPs which differentiates them from AMPs. Due to the similar Arg-rich structural motif, CPPs and AMPs have strong Arg-lipid and Arg-water interactions, which stabilize these hydrophobic peptides by membrane neutralization and water solvation and thus facilitate the insertion. However, CPPs interact with the cellular membrane in a non-invasive manner, while AMPs function by disrupting the lipid membrane. In addition, CPPs also experience charge–charge interaction from the distal phosphate layer and the guanidinium–phosphate interactions stabilize the CPP peptides in lipids and facilitate the insertion, while the plastic conformation and high mobility further promote the translocation.
For AMPs that preferentially attack internal cellular targets, similar translocation mechanisms have been reported: for buforin 2, which translocates efficiently, but with little membrane activity [29], the structure and orientation in the bilayer have been observed to be very similar to those of magainin 2 [30]. From these results a model was proposed whereby buforin 2 molecules would form a toroidal pore, just as magainin 2 does, but less stable; this would result in shorter pore lifetimes—with a concomitant decrease in permeabilization—at the same time that the translocation rate would increase because pore disintegration, which is the actual translocation step, would become more frequent [29, 30]. This model is supported by results that show that the presence of bilayer components that prevent the formation of toroidal pores (such as dioleyl phosphatidylethanolamine [31]) inhibit buforin 2 translocation, whereas anionic phospholipids, which decrease the charge repulsions between the cationic peptide molecules, stabilize the pore to a point that significant leakage and flip-flop is observed [30]. Buforin 2 translocation has also been shown to withstand cargo addition, as demonstrated by the attachment of green fluorescent protein [32], which makes this peptide a promising candidate for its development into a CPP. A “membrane-thinning” effect was proposed for the AMP magainin 2 [33], in which the peptide aggregates on the surface of the membrane and the decreased local surface tension allows the peptide to intercalate the membrane.
3 Antimicrobial Peptides
Antimicrobial peptides are gene-encoded, ribosomally synthesized polypeptides. They usually have common characteristics: small peptide with a varying number (from five to over a hundred) of amino acids, strongly cationic (pI 8.9–10.7), heat-stable (100 °C, 15 min), no drug fastness and no effect on eukaryotic cells [34]. In total, more than 5000 AMPs have been discovered, predicted or synthesized up to date [35]. The natural AMPs have been isolated and characterized and produced from practically all living organisms, ranging from prokaryotes (e.g. bacteria) to eukaryotes (e.g. protozoan, fungi, plants, insects, and animals) [36, 37]. In animals, AMPs are mostly found in the tissues and organs that are exposed to airborne pathogens and are believed to be the first line of the innate immune defense [38] against viruses, bacteria, and fungi [37]. Several types of eukaryotic cells are involved in AMP production such as lymphs, epithelial cells in gastrointestinal and genitourinary systems [39], phagocytes [40], and lymphocytes of the immune system [37]. In addition to direct involvement in innate immunity, AMPs have also been found to influence inflammatory responses during an infection [41].
In short, AMPs have the ability to kill pathogenic microorganisms, including gram-positive and gram-negative bacteria, viruses, protozoa, and fungi. In contrast to conventional antibiotics, AMPs appear to be bactericidal (kills bacteria) instead of bacteriostatic (inhibits growth). They can destroy bacteria within minutes with the rate being faster than the bacteria growth rate [42].
3.1 Origins and Classification of AMPs
3.1.1 Origin of AMPs
AMPs can be commonly classified into four groups according to their origins. They can originate from insects, other animals, synthesis, and genetically engineered microorganisms. It is possible to make fully synthetic peptides by chemical synthesis [43] or by using recombinant expression systems [44]. These artificial sources of AMPs are useful for the modification of existing AMPs and for designing new synthetic AMPs. Such modifications have potential to change the targets of AMPs and improve the stability of AMPs against proteases [45]. Despite these advantageous features of AMPs, there are still some challenges to their applications, such as potential toxicity to humans [46], sensitivity to harsh environmental conditions (susceptibility to proteases and extreme pH) [47], lack of selectivity against specific strains [48], high production costs [49], folding issues of some large AMPs [50], reduced activity when used for surface coating, and bacterial resistance to some AMPs [51].
Historically, AMPs have also been referred to as cationic host defense peptides [52], anionic antimicrobial peptides/proteins [53], cationic amphipathic peptides [54], cationic AMPs [55], host defense peptides [56], and α-helical antimicrobial peptides [57]. The discovery of AMPs dates back to 1939, when Dubos extracted an antimicrobial agent from a soil Bacillus strain. This extract was demonstrated to protect mice from pneumococci infection. In the following year, Hotchkiss and Dubos fractionated this extract and identified an AMP which was named gramicidin [58]. In 1941, another AMP, tyrocidine, was discovered and found to be effective against both gram-negative and gram-positive bacteria. However, tyrocidine exhibited toxicity to human blood cells. In the same year, another AMP was isolated from a plant Triticumaestivum, which was later named purothionin and found effective against fungi and some pathogenic bacteria [58]. The first reported animal-originated AMP is defensin, which was isolated from rabbit leukocytes in 1956. In the following years, bombinin from epithelia and lactoferrin from cow milk were both described. During the same time, it was also proven that human leukocytes contain AMPs in their lysosomes [58].
3.1.2 How Do AMPs Get Inside Cells?
There is no common molecular entry route for AMPs—it depends on the nature of the peptide, the membrane lipid composition and the peptide/lipid ratio. The mechanism comprises several stages which are not yet fully understood, despite extensive studies. The necessary step is peptide’s association with membrane lipids which results in long-range defects. The different molecular mechanisms postulated (such as barrel-stave or toroidal/wormhole pore formation, aggregate channel formation or surfactant-like interactions [59]) assume that aggregation/oligomerization of AMP in the cytoplasmic membrane is the necessary step leading to the membrane lysis.
3.1.3 Classification
Most AMPs reported to date can be classified based on one of the following four types based on their secondary structural features: such as cathelicidins (with a linear α-helical structure), defensins (with a β-strand structure), and bactenecins (with a loop structure) [60], and extended helices with a predominance of one or more amino acids. Among these structural groups, α-helix and β-sheet structures are more common [61] and α-helical peptides are the most studied AMPs to date. The best known examples of such AMPs are protegrin, magainin, cyclic indolicin, and coiled indolicin [57] and α-helical peptides without the presence of cysteines in the sequence, such as melittin [62]. β-Sheet peptides are composed of at least two β-strands with disulfide bonds between these strands [63], such as the protegrins [64]. And the AMPs with intermolecular disulfide bonds exhibiting loop/hairpin-like structures, such as bactenecin [65], belong to the third group. The final groups are peptides with predominance of one or more distinct amino acids, such as the proline/arginine-rich peptide Bac7 [66].
3.1.4 AMP Design
The design of novel AMPs requires consideration of several factors, including secondary structure, amphipathicity, and the presence of positively charged residues. It is believed that an amphipathic secondary structure is required in order for AMPs to function, although the exact mechanism of action is still unclear. As pointed out by Tossi et al. [67], the design of AMPs is generally based on: (a) amino acid residue analogues of natural peptides (e.g. congeners) that differ at one or more residue positions, are shortened or contain deletions, as well as hybrid AMPs composed of fragments of two different natural peptides; (b) amino acid residues that maximize the amphipathic nature of AMPs; (c) amino acid sequences from combinatorial libraries; and (d) amino acid sequences that are patterned from known, naturally occurring, α-helical peptide domains.
Important factors to consider when designing AMPs are the length of the peptide [68], net charge [69, 57], amphipathicity [70], and possible modifications such as phosphorylation [71], addition of d-amino acids [72], methylation [19], amidation [73], glycosylation [74], formation of disulfide linkage [75], and proteolytic cleavage [76].
3.2 Mechanisms of Action of AMPs
It is generally accepted that positively charged peptides interact directly with the negatively charged cellular membranes of bacterial cells, resulting in the increase of membrane permeability, which leads to a rapid cell death [77]. The groups of AMPs can be divided as: (a) antibacterials; (b) antivirals; (c) antifungals; (d) antiparasitics; and (e) anticancer peptides. AMPs kill bacteria by inhibiting some important pathways inside the cell such as DNA replication and protein synthesis [78]. Antiviral AMPs neutralize viruses by integrating in either the viral envelope or the host cell membrane. AMPs can integrate into viral envelopes and cause membrane instability, rendering the viruses unable to infect host cells [79]. AMPs can also reduce the binding of viruses to host cells [80]. Some of antifungal peptides are capable of binding to chitin. Such binding ability helps AMPs to target fungal cells efficiently. Cell wall-targeting antifungal AMPs kill the target cells by disrupting the integrity of fungal membranes by increasing permeabilization of the plasma membrane [81], or by forming pores directly [82]. An example of antiparasitic peptide is cathelicidin, which is able to kill Caernohabditis elegans by forming pores in the cell membrane [83]. Even though some parasitic microorganisms are multicellular, the mode of action of antiparasitic peptides is the same as other AMPs. They kill cells by directly interacting with cell membrane [83]. Anticancer peptides are also known as host defense peptides. They unction by targeting the cell membrane, as to date, more than 100 host defense peptides are known.
3.3 Applications of AMPs
Antimicrobial peptides represent a novel class of therapeutic agents that may be useful in the treatment of a range of infectious diseases. However, in order to develop antimicrobial peptides for therapeutic use, there are a number of technological hurdles to address, including optimizing peptide stability and antimicrobial activity. Most pharmaceutical efforts has been devoted to the development of topically applied agents, such as magainin analog pexiganan, largely because of the relatively safety of topical therapy and the uncertainty surrounding the long-term toxicology of any new class of systemically administrated drug. Diverse applications have been demonstrated for antimicrobial peptides as anti-infective agents. The broad antimicrobial spectrum of antimicrobial peptides positions them for consideration as “chemical condoms” to limit the spread of sexually transmitted diseases, including Neisseria, Chlamydia, human immunodeficiency virus (HIV), and Herpes simplex virus (HSV).
3.3.1 AMP-Based Drug Development
In addition to the previously mentioned applications of AMPs, there are much broader uses for the peptides. This is a subject that has been previously thoroughly reviewed by several authors [84–86], therefore only the select few approaches that the authors regarded to be of interest will be discussed in the section at hand.
One noteworthy example entails the CPP-PMO conjugate developed in cooperation with AVI Biopharma. The AVI-6002 and AVI-6003 molecules developed by them show promising results against the mRNA translation process of Ebola virus and Marburg virus, which becomes increasingly important in the light of the recent Ebola outbreak. Various in vivo studies have been conducted with these drug candidates, including mice, guinea pigs and primates [87, 88]. Recently, they have been conducting preliminary clinical trials as well and have obtained positive results from phase 1 trials [88].
Another molecule of interest is a cationic peptide-based drug targeting fungal infections on toenails, is showing promise and has recently entered clinical studies. Novabiotics has created a cyclic hepta-arginine antimicrobial peptide that shows potency in inhibiting fungal growth by disrupting cellular membranes, leading to the loss of viability in affected fungal cells. The cyclic form that was introduced to the molecule is claimed to enhance the antifungal capabilities of the peptide and to increase its stability [89].
3.3.2 CPPs with AMP Properties
In recent years it has been discovered that the Tat peptide shows potent antibacterial activity (MIC 2–8 μM) against a broad spectrum of pathogens including gram-positive and gram-negative bacteria such as S. aureus and also fungi such as Saccharomyces cerevisiae and Candida albicans [90, 91]. The peptide internalizes without any damage to the cell membrane, thus being cytotoxic inside the cells, leading for example to a fast accumulation in the nucleus in fungi, where it causes cell cycle arrest in G1 phase. The uptake of the Tat peptide seems to be sequence-dependent and not induced by its secondary structure [92]. Nearly 20 years after its discovery, it was observed that the CPP penetratin is a potent antimicrobic against gram-negative and gram-positive bacteria such as Bacillus megaterium. An MIC of 0.5–4 μM was measured, and the peptide showed no cytotoxicity against mammalian cells.
In the 18-amino-acid peptide pVEC, uptake takes place by a non-endocytotic mechanism of translocation without alteration of plasma membrane permeability or cell morphology. After the internalization process, it is mainly localized in nuclear structures and was used as a carrier for peptide nucleic acids (PNAs) and proteins [93]. It can enter mammalian and microbial cells and preferentially permeates and kills microbes; for example, it was described to killMycobacterium smegmatis at low micromolar doses at which normal human cells were not damaged [94].
Pep-1 has a hydrophobic tryptophan-rich domain, a spacer domain, and a hydrophilic lysine-rich domain and is characterized by an amphipathic α-helical structure [95]. Pep-1 has a broad antimicrobial spectrum against gram-negative and gram-positive bacterial strains but weak antibacterial activity [96]. A bacteria-selective variant could be designed by replacing several glutamic acids with lysines. The modified peptide showed high activity (MIC 1–2 μM) against strains of gram-positive and gram-negative bacteria as well as against clinical isolates of multidrug-resistant Pseudomonas aeruginosa (MDRPA) and methicillin-resistant S. aureus (MIC 1–8 μM) [96]. Certain chimeric peptides, such as Pep-1, may even be considered a “blend” between AMPs and CPPs. Although reported as a CPP, Pep-1 is a strongly amphipathic cationic peptide, rich in basic amino acids and tryptophan, having a proline residue in its sequence. These are the classical characteristics attributed to AMPs. The ability to cysteine-bridge monomers, which greatly improves translocation efficiency, further increases the similarities to AMPs. Not surprisingly, Pep-1 uses mainly physical routes to translocate membranes. However, this route is not always operative [95] and endocytic pathways are alternatives.
Transportan (TP) is a 27 amino acid chimeric peptide composed of the neuropeptide galanin and mastoparan-X linked by a lysine. It exhibits rapid and non-endocytotic uptake and was used for delivery of peptides, PNA oligomers or even intact proteins [97]. TP 10 is a 21-amino-acid deletion analog of the chimeric CPP transportan that contains the mastoparan sequence but lacks the toxicity of its parent compound. It can enter mammalian and microbial cells and preferably permeate and kill microbes such as S. aureus at low micromolar doses but does not damage human cells [98].
The cationic and amphipathic model peptide (MAP) is another CPP with antimicrobial properties. The uptake of the peptide again seems to be a combination of energy-dependent and energy-independent mechanisms, whereas the amphipathicity of the peptide is crucial for uptake [99]. It exhibits an antimicrobial effect against gram-positive bacteria such as B. megaterium and gram-negative bacteria such as E. coli in a low micromolar range. However, no antifungal activity against yeast S. cerevisiae has been observed [94]. A pore-formation mechanism was proposed for MPG (a 27-residue amphipathic peptide) and Pep-1 [100], which is also a common mechanism used by antimicrobial peptides (Table 1).
Increasing evidence indicates that membrane-interacting peptides, in fact, may exhibit cross-functionality, e.g. some AMPs possess the ability to cross mammalian cell membranes by non-damaging processes, while several CPPs display significant antimicrobial activity. For example, Tat48–60 from the HIV-1 transactivating protein has been shown to inhibit growth of both gram-positive and gram-negative bacteria as well as of fungi [90, 91, 101]. Similarly, Pep-1 derived from simian virus has been modified (Glu → Lys) into an antimicrobial analog, Pep-1-K that possesses activity toward gram-positive and gram-negative bacteria [102]. Also, pVEC derived from cadherin exhibits activity toward both gram-negative and gram-positive bacteria [94, 98], while TP10, a deletion analog of the chimeric CPP transportan, possesses potency against gram-positive bacteria and fungi [98]. Furthermore, the designed model amphipathic peptide (MAP) shows antibacterial activity, whereas it does not exhibit antifungal activity [94].
4 Antiviral Peptides
Antiviral peptides form a distinct subcategory of antimicrobial peptides. While they share many common features with other antimicrobial peptides targeted against bacteria, fungi, and other microorganisms, these peptides, which are capable of inhibiting viruses, should be discussed separately. Unlike other microorganisms, that can be classified as living, viruses fall somewhere between living and non-living categories. Although they possess the capability to replicate, viruses do not have a metabolism and they lack the means to produce energy like living things do with the help of mitochondria. Instead, they recognize host cells, after which the execution of gene expression, genome replication, and virion formation will take place [103]. This is the main reason why antiviral substances can be considered a class of their own—as viruses are so varied, the drug which might show great promise against one type of virus is, with great probability, completely inefficient against some other virus, in some cases this lack of effect can even be observed against a different subtype of the same virus (a common example is HIV-1 with its various strains) [104, 105]. Furthermore, when the main focus of antibacterial, antifungal or even antitumor peptides is to neutralize the organism/cell that they have come to interact with, antiviral peptides must be capable of disabling the viral infection in such a way that the host cell would remain intact and operational, otherwise the host might be weakened by a great extent and, in worst cases, even suffer terminal organ failures.
4.1 CPPs with AVP Properties
While the main antiviral approach has been the use of a CPP-drug conjugate, some CPPs have demonstrated to possess antiviral properties by themselves. One of the viruses against which the antiviral effect of CPPs is evident is HIV-1. It has been described by Keogan et al. that a HIV-1-derived CPP, the Tat-peptide, interacts with CXCR4 and subsequently inhibits the replication efficiency of the virus strains that use CXCR4 as their co-receptor [105]. Another successful approach is to modify the antiviral molecule to achieve cell-penetrating capabilities, like a direction taken by Zhang et al., in the case of which they used hydrocarbon stapling to create a more stable secondary structure and enhance the a-helicity of the peptide [106]. Following the success of the first hydrocarbon-stapled peptide, the research group has produced a wide range of such antiviral CPPs known as NYAD peptides [107, 108].
Antiviral CPPs can also be created by a synthetic approach, which is best illustrated by the creation of synthetic LK peptides [109]. The reason for such naming is that they are composed either only or mainly of leucine (L) and lysine (K) residues. The key elements in achieving the enhanced antiviral efficiency were once again the increased a-helicity and the amphipathic characteristics of the molecules, which further stresses the importance of these traits when developing antiviral CPPs. Some synthetic antiviral CPPs can also be a result of a lucky accident. While trying to improve the efficiency of their CPP-PNA conjugate [110] by adding a fatty acid domain, they created a CPP, which was an efficient antiviral agent even without the conjugated PNA. The infected cells treated with the peptide also demonstrated the tendency to release naked viral nucleocapsids that are extremely immunogenic, which could be used for vaccine-like immune system stimulation in the infected organism [111].
Screening of phage display libraries is also a possibility when looking for antiviral agents with penetrating properties. The use of this approach by Tiwari et al. has yielded peptide groups G1 (possesses alternating charges) and G2 (possesses repetitive charges) that bind heparin sulfate and inhibit viral entry by attaching themselves to cell surface and blocking the virus–cell interactions and membrane fusions [112]. While the main approach of the study was the prophylactic treatment against HSV-1 on the surface of the cell, it was noted that the particle is possibly internalized, in which case it can be classified as a CPP. This is further confirmed by additional study, where G2 has been conjugated to acyclovir in order to increase its antiviral efficiency [113]. And while the cell penetration might not prove to be especially relevant in the antiviral content of this study, it shows that the screening methodology contains in itself the capability to identify other peptides that possess cell-penetrating features.
5 Therapeutic and Scientific Applications of CPPs in the Field of Microbes and Viruses
5.1 Application of AMPs as Drug Delivery Vectors
As previously mentioned, AMPs have been described to show CPP properties as well, therefore they can be used as delivery vectors for several therapeutic and diagnostic molecules in the treatment of cancer, genetic, cardiovascular, inflammatory, and infectious diseases. For example, pyrrhocoricin consists of 20 amino acids and is an antimicrobial peptide isolated from insects, being effective against gram-negative bacteria but almost inactive against gram-positive strains [114]. Furthermore, pyrrhocoricin itself has been used as a drug delivery system. Otvos et al. investigated a modified pyrrhocoricin dimer that could successfully deliver peptide antigens into dendritic cells and human fibroblasts [115]. Another example is the antimicrobial protein lactoferrin (hLF), the human milk protein, which is a very important protein in immune defense due to its antifungal, antimicrobial, and antiviral activities [116]. The truncated version of this peptide consists of 49 amino acids and exhibits antimicrobial, antiviral, antitumor, and immunological activity [117] and was described to enter different cells efficiently (e.g. Hela or rat IEC-6). The uptake mechanism of the hLF peptide seems to be concentration-dependent and for concentration higher than 10 μM, rapid delivery into the cytoplasm and nucleus is observed. Furthermore, the uptake pathway was determined to be sensitive to rottlerin, a protein kinase inhibitor with specificity for protein kinase C (PKC). This was also observed for the uptake of arginine-rich CPPs such as Tat and R9 [118]. The uptake efficiency is supposed to be conformation-dependent, because the cyclic structure is required for binding to heparin sulfate and correlates with lipid-induced conformational changes. Several examples of efficient cargo delivery have been described for the hLF peptide; especially proteins and high-molecular-weight complexes have been successfully transported [119].
Another peptide which combines cell-permeation and antimicrobial properties is Bactenecin 7 (Bac7) [120]. This is a linear 59-residue protein that was isolated from large granules of bovine neutrophils. Bac7 belongs to the bactenecin family and consists of four 14-residue repeats. It also belongs to the Pro/Arg-rich family and was described to be antimicrobially active against gram-negative bacteria in a micromolar range but not against gram-positive strains [121]. The antimicrobial effect is caused by inhibition of the intracellular protein synthesis machinery in a two-step mechanism, where the first is entry of the peptide into the cytoplasm and the second is intracellular inhibition of its target [120]. Generally, the longer segments of Bac-7, containing antibacterial and intracellular delivery regions, have antibacterial and cell-permeating activity. Also, SynB vectors from the antimicrobial peptide protegrin-1 (PG-1) can be used for cargo delivery purposes. It shows potent activity against fungi, bacteria, and several enveloped viruses. Like other AMPs, it interacts with the lipid matrix of bacterial membranes and forms pores [75].
The replacement of four cysteines and two valines of PG-1 led to linear peptides (SynB1) still able to penetrate cells efficiently but without being cytolytic to them. With the help of this, peptide transport of covalently coupled doxorubicin to the brain has been reported. The blood–brain barrier was crossed with high efficiency and without any compromise to its integrity [122]. Another example is buforin II, a 21-amino-acid antimicrobial peptide that was discovered in stomach tissue of Asian toad. It penetrates through the cell membrane without destroying it and kills bacteria by binding to nucleic acids. BF2d, which is a modified analog of buforin II, exhibits cell-penetrating properties and is able to deliver the GFP protein to HeLa cells [32]. Recently, a cathelicidin-derived carrier peptide, sC18, has been developed. This peptide originates from the 18-kDa cationic antimicrobial protein (CAP18) that was first isolated from rabbit leukocytes. Like the hLF protein, CAP18 is a lipopolysaccharide binding protein with an α-helical structure [123]. CAP18 itself and also shortened variants exhibit antimicrobial properties in the lower micromolar range [124].
5.2 CPPs as Antiviral Drug Delivery Vectors
The delivery of antiviral drugs into eukaryotic cells has become increasingly popular over the years. While the specific therapeutic strategies to inhibit viruses might vary, it can be claimed that nearly all CPP-drug combinations, with a select few exceptions, make use of covalent bonding, effectively creating a single molecule which is efficient both in cellular penetration and in its antiviral activity with the advantage of being non-cytotoxic in a wide selection of cell lines and in vivo environments (w6). Here, we present a short overview of the studies that have successfully applied such fusion peptides in their research.
A common approach for antiviral treatment is the use of PNA-CPP conjugates. This strategy has been described to target such high-profile viruses such as HIV-1 and HCV with efficiency up to 99 % [125, 126]. The CPPs commonly used as a backbone for such molecules are Tat, Transportan, and Penetratin [127, 126, 128]. While the main target of the research has been virus inhibition in the cell culture environment, Ganguly et al. have demonstrated efficient uptake and slow clearance levels in Balb/C mice, suggesting a potential therapeutic application for their conjugate [127]. Another interesting result was produced by Chaubey et al., who described that a Transportan-PNA molecule, which had previously been shown to display significant antiviral efficiency (14a), is also a potent virucidal agent, rendering HIV-1 virions pre-treated with the conjugate noninfectious, suggesting a probable prophylactic medical application for their molecule [129]. PNA-CPPs such as the Tat-FS can also be used to block the frameshift process required for the replication of such viruses as SARS-Coronavirus, making them a viable tool against any viruses that require frameshifting to take place during their replication [130]. A successful Tat-based application of PNAs has been shown to lead to Japanese encephalitis virus inhibition by competing with RdRp through interactions with its binding sites at 3′ regions of the RNA [131].
An interesting anti-HIV-1 development has been published by Zhuang et al. Their approach was to combine an antibody fragment with the Tat-peptide, suppressing the reverse transcriptase activity of CCR5-topic HIV-1 isolates in primary blood mononuclear cells. What makes this development especially interesting is the fact that the anti-Rev antibody fragment did not have any antiviral activity on its own and only obtained the capability for strong inhibition after being conjugated to the Tat-peptide [104]. Penetratin has also been used to deliver antibody-derived antiviral agents known as humanized-VH/VHH, which displayed strong helicase binding affinity in order to inhibit HCV replication [132]. Furthermore, single-chain variable fragment antibodies have been attached to cytoplasmic transduction peptide in order to target the HBV core protein and to inhibit the nucleocapsid assembly and replication processes of the virus [133].
Mino et al. have reported the creation of cell-permeable artificial zinc-finger proteins in order to reduce HPV replication levels. Their approach entailed fusing their protein to the polyarginine CPP R9, allowing to block Rep binding to its replication origin sequence [134]. Proteins such as Mx-1 have been used to inhibit VSV virus in combination with Tat peptide through the suppression of replication and capability to cure ongoing infections [135]. Hexa-arginine with a synthetic peptide mimicking a β-sheet/loop motif commonly found in the C-terminal domain of the Chandipura virus P protein binds the positive-sense leader RNA to inhibit virus replication [136]. Heat shock protein gp96 has been fused to Tat-peptide in an attempt to enhance T-cell-based immune responses. This led to increased antiviral and antitumor potency and to the inhibition of HBV in mice [137]. Protease-inhibiting peptides have been developed against HCV infection. These molecules were conjugated to a selection of CPPs and the fusion with Antennapedia was regarded as the approach with the highest efficiency [138]. Peptide-targeting HIV-1 capsid formation has been successfully applied using a commercial CPP-based transfection reagent, Chariot. What makes this development especially interesting is that this is one of the rare occasions upon which the molecule of interest is not conjugated to the transfection peptide covalently but employs non-covalent bonding strategy instead [139]. The approach to target the capsid of the virus has also been used in HCV inhibition—the capsid has been targeted by R7 in fusion with nucleocapsid binding subunits. In addition to its inhibition of envelopment, the peptide conjugate also blocks the subsequent viral particle release [140].
A less common approach is to conjugate a siRNA molecule to a CPP, as it was done by Meng et al. using Tat-1 as a peptide backbone. Their siRNA designed against the 5′ UTR of HCV managed to efficiently enter the cells and inhibit the replication of viral RNA [141]. PMOs have been utilized in an approach against murine hepatitis virus, which is a member of coronavirus family which holds such other, better known members, such as SARS. Various arginine-rich PPMO combinations were compared and it resulted in the selection of (R-Ahx-R)4AhxB–PPMO over R9F2C–PPMO and Tat-PPMO based on their delivery efficiency in a splice-correcting model system [142] and antiviral efficiencies targeted against 5′-terminal nucleotides of the viral genome [143]. The P007 CPP-conjugated PMO targeting the 3′ region of the internal ribosomal entry site was shown to hold great potency against the coxsackievirus B3, a known cause for viral myocarditis, both in cell culture and in mice, showing a perspective for further therapeutic research [144]. PMOs in combination with arginine-rich CPPs, such as AVI-6002 and AVI-6003 have also been reported to be capable of inhibiting Ebola and Marburg viruses respectively. The main mechanism of action for these molecules is the inhibition of viral mRNA translation, which leads to the inhibition of the whole replication process [88].
5.3 Use of CPPs in Antiviral Research
While there are a large number of possibilities present to use CPPs to combat viral infections, some applications for such peptides can, instead, help the researchers to understand the virus more thoroughly. While it may not be apparent at first, there are possible applications present for almost any type of CPP when it comes to fundamental research. If to consider, for example, a CPP delivery vector that has been proven to have no effect whatsoever in regards to viral infection-replication cycle and also shows no considerable cytotoxic effects against the host cell, such as the CPP PepFect6 when combined with Semliki Forest Virus [145], a member of alphavirus family, it opens up numerous possibilities to gain insight into the properties of the virus and about its behavior when some molecule of interest has been introduced into the environment. This lack of interference becomes especially important when taking into account the fact that other widely used transfection methods, such as electroporation [146] or lipofection [147] either damage the cells to a great extent or inhibit the infection and replication capabilities of viruses [145], resulting in data about the virus that can be considered as unreliable.
But the research applications of CPPs are not limited to the experimental set-ups where the virus is left undisturbed. As mentioned before, there are numerous occasions where CPP-based vector has been used to deliver an antiviral substance into the cells. This approach, combined with the fact that some CPPs do not affect viruses, allows the conduction of research related to viral replication. If to introduce a CPP-drug conjugate into the cellular environment that completely inhibits late-stage virion formation, it could create a setting in which it is easier to observe viral replication without the need to create replicon models of the original virus, the downside of which is that while they do carry out the replication in a similar way to the virus of interest, they are still, in essence, a shortened version of the original genome, resulting in a possible change of replication speeds and cellular immune response [148]. In the case of highly mutagenic viruses, such as HIV-1, it would also open up the possibility to efficiently research the abilities of the viruses to overcome some obstacle in their life cycle—for example which beneficial mutations is the virus capable of obtaining if a certain replication stage or pathway is blocked. Similar capability has been observed by Zhang et al., where they observed mutations in viral proteins to overcome the antiviral effect of their CPP molecules [107].
Another interesting research topic is the intracellular detection of viruses. CPP vectors have successfully been used to transport small molecular beacons into eukaryotic cells with the goal to observe the behavior of viruses in real-time. A good example is the experiment by Yeh et al. in which they describe using the widely used Tat-peptide to transport nuclease-resistant molecular beacons to detect coxsackievirus B6 replication [149]. They used this approach to verify the presence of viral RNA in cells and were able to observe the spreading of replication from one cell to another with great accuracy. Small molecule transport can be used to identify the various interactions that certain virus regions might have with the host cell. Caignard et al. used a Tat-conjugated peptide version of the measles virus V protein to characterize in which way do the intracellular interactions between the virus and IFN-α/β signaling pathway take place [150].
6 Conclusion
Antimicrobial peptides prove themselves to be highly efficient and versatile therapeutic systems that allow the inhibition of various pathogens. With the advent of CPPs, the two fields are evolving closely, resulting in various combinations, adoptions of antimicrobial strategies and novel approaches. As demonstrated, the line between AMPs and CPPs can be very thin at times, with several molecules displaying characteristics common to both groups. In addition, CPPs can be made to serve antimicrobial or fundamental research purposes either by themselves or in conjugation with various inhibitory molecules, further increasing the spectrum of this field.
References
Järver P, Mäger I, Langel Ü (2010) In vivo biodistribution and efficacy of peptide mediated delivery. Trends Pharmacol Sci 31:528–535
Lindgren M, Rosenthal-Aizman K, Saar K et al (2006) Overcoming methotrexate resistance in breast cancer tumour cells by the use of a new cell-penetrating peptide. Biochem Pharmacol 71:416–425
Santra S, Yang H, Stanley JT et al (2005) Rapid and effective labeling of brain tissue using TAT-conjugated CdS:Mn/ZnS quantum dots. Chem Commun (Camb) 3144–3146
Lewin M, Carlesso N, Tung CH et al (2000) Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat Biotechnol 18:410–414
Vasconcelos L, Pärn K, Langel Ü (2013) Therapeutic potential of cell-penetrating peptides. Ther Deliv 4:573–591
Vives E, Schmidt J, Pelegrin A (2008) Cell-penetrating and cell-targeting peptides in drug delivery. Biochim Biophys Acta 1786:126–138
Nakase I, Akita H, Kogure K et al (2011) Efficient intracellular delivery of nucleic acid pharmaceuticals using cell-penetrating peptides. Acc Chem Res 45:1132–1139
Koren E, Torchilin VP (2012) Cell-penetrating peptides: breaking through to the other side. Trends Mol Med 18:385–393
Mayor S, Pagano RE (2007) Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol 8:603–612
Futaki S, Nakase I, Tadokoro A et al (2007) Arginine-rich peptides and their internalization mechanisms. Biochem Soc Trans 35:784–787
Richard JP, Melikov K, Brooks H et al (2005) Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem 280:15300–15306
Vandenbroucke RE, De Smedt SC, Demeester J et al (2007) Cellular entry pathway and gene transfer capacity of TAT-modified lipoplexes. Biochim Biophys Acta 1768:571–579
Richard JP, Melikov K, Vives E et al (2003) Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem 278:585–590
Su Y, Waring AJ, Ruchala P et al (2010) Membrane-bound dynamic structure of an arginine-rich cell-penetrating peptide, the protein transduction domain of HIV TAT, from solid-state NMR. Biochemistry 49:6009–6020
von Heijne G (2006) Membrane-protein topology. Nat Rev Mol Cell Biol 7:909–918
Brogden KA, Ackermann M, Huttner KM (1997) Small, anionic, and charge-neutralizing propeptide fragments of zymogens are antimicrobial. Antimicrob Agents Chemother 41:1615–1617
Lai R, Liu H, Hui Lee W et al (2002) An anionic antimicrobial peptide from toad Bombina maxima. Biochem Biophys Res Commun 295:796–799
Steffen H, Rieg S, Wiedemann I et al (2006) Naturally processed dermcidin-derived peptides do not permeabilize bacterial membranes and kill microorganisms irrespective of their charge. Antimicrob Agents Chemother 50:2608–2620
Hancock RE, Chapple DS (1999) Peptide antibiotics. Antimicrob Agents Chemother 43:1317–1323
Zasloff M (2002) Antimicrobial peptides of multicellular organisms. Nature 415:389–395
Tossi A, Sandri L, Giangaspero A (2000) Amphipathic, alpha-helical antimicrobial peptides. Biopolymers 55:4–30
Park KH, Nan YH, Park Y et al (2009) Cell specificity, anti-inflammatory activity, and plausible bactericidal mechanism of designed Trp-rich model antimicrobial peptides. Biochim Biophys Acta 1788:1193–1203
Caesar CE, Esbjorner EK, Lincoln P et al (2006) Membrane interactions of cell-penetrating peptides probed by tryptophan fluorescence and dichroism techniques: correlations of structure to cellular uptake. Biochemistry 45:7682–7692
Adao R, Nazmi K, Bolscher JG et al (2011) C- and N-truncated antimicrobial peptides from LFampin 265–284: biophysical versus microbiology results. J Pharm Bioallied Sci 3:60–69
Hong M, Su Y (2011) Structure and dynamics of cationic membrane peptides and proteins: insights from solid-state NMR. Protein Sci 20:641–655
Tang M, Hong M (2009) Structure and mechanism of beta-hairpin antimicrobial peptides in lipid bilayers from solid-state NMR spectroscopy. Mol Biosyst 5:317–322
Zhang Y, Doherty T, Li J et al (2010) Resonance assignment and three-dimensional structure determination of a human alpha-defensin, HNP-1, by solid-state NMR. J Mol Biol 397:408–422
Ulmschneider MB, Ulmschneider JP (2008) Membrane adsorption, folding, insertion and translocation of synthetic trans-membrane peptides. Mol Membr Biol 25:245–257
Kobayashi S, Takeshima K, Park CB et al (2000) Interactions of the novel antimicrobial peptide buforin 2 with lipid bilayers: proline as a translocation promoting factor. Biochemistry 39:8648–8654
Kobayashi S, Chikushi A, Tougu S et al (2004) Membrane translocation mechanism of the antimicrobial peptide buforin 2. Biochemistry 43:15610–15616
Matsuzaki K, Murase O, Fujii N et al (1995) Translocation of a channel-forming antimicrobial peptide, magainin 2, across lipid bilayers by forming a pore. Biochemistry 34:6521–6526
Takeshima K, Chikushi A, Lee KK et al (2003) Translocation of analogues of the antimicrobial peptides magainin and buforin across human cell membranes. J Biol Chem 278:1310–1315
Ludtke S, He K, Huang H (1995) Membrane thinning caused by magainin 2. Biochemistry 34:16764–16769
Li Y, Xiang Q, Zhang Q et al (2012) Overview on the recent study of antimicrobial peptides: origins, functions, relative mechanisms and application. Peptides 37:207–215
Zhao X, Wu H, Lu H et al (2013) LAMP: a database linking antimicrobial peptides. PLoS One 8:e66557
Conlon JM, Sonnevend A (2010) Antimicrobial peptides in frog skin secretions. Methods Mol Biol 618:3–14
Radek K, Gallo R (2007) Antimicrobial peptides: natural effectors of the innate immune system. Semin Immunopathol 29:27–43
Schauber J, Gallo RL (2008) Antimicrobial peptides and the skin immune defense system. J Allergy Clin Immunol 122:261–266
Ganz T (2003) The role of antimicrobial peptides in innate immunity. Integr Comp Biol 43:300–304
Hancock RE, Scott MG (2000) The role of antimicrobial peptides in animal defenses. Proc Natl Acad Sci U S A 97:8856–8861
Kindrachuk J, Jenssen H, Elliott M et al (2013) Manipulation of innate immunity by a bacterial secreted peptide: lantibiotic nisin Z is selectively immunomodulatory. Innate Immun 19:315–327
Lohner K (2009) New strategies for novel antibiotics: peptides targeting bacterial cell membranes. Gen Physiol Biophys 28:105–116
Wade JD, Lin F, Hossain MA et al (2012) Chemical synthesis and biological evaluation of an antimicrobial peptide gonococcal growth inhibitor. Amino Acids 43:2279–2283
Ramos R, Moreira S, Rodrigues A et al (2013) Recombinant expression and purification of the antimicrobial peptide magainin-2. Biotechnol Prog 29:17–22
Papo N, Oren Z, Pag U et al (2002) The consequence of sequence alteration of an amphipathic alpha-helical antimicrobial peptide and its diastereomers. J Biol Chem 277:33913–33921
Matsuzaki K (2009) Control of cell selectivity of antimicrobial peptides. Biochim Biophys Acta 1788:1687–1692
Svenson J, Stensen W, Brandsdal BO et al (2008) Antimicrobial peptides with stability toward tryptic degradation. Biochemistry 47:3777–3788
Eckert R, Qi F, Yarbrough DK et al (2006) Adding selectivity to antimicrobial peptides: rational design of a multidomain peptide against Pseudomonas spp. Antimicrob Agents Chemother 50:1480–1488
Bommarius B, Jenssen H, Elliott M et al (2010) Cost-effective expression and purification of antimicrobial and host defense peptides in Escherichia coli. Peptides 31:1957–1965
Duquesne S, Destoumieux-Garzon D, Zirah S et al (2009) Post-translational modification and folding of a lasso-type gene-encoded antimicrobial peptide require two enzymes only in Escherichia coli. Adv Exp Med Biol 611:35–36
Marr AK, Gooderham WJ, Hancock RE (2006) Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr Opin Pharmacol 6:468–472
Brown KL, Hancock RE (2006) Cationic host defense (antimicrobial) peptides. Curr Opin Immunol 18:24–30
Harris F, Dennison SR, Phoenix DA (2009) Anionic antimicrobial peptides from eukaryotic organisms. Curr Protein Pept Sci 10:585–606
Groenink J, Walgreen-Weterings E, van’t Hof W et al (1999) Cationic amphipathic peptides, derived from bovine and human lactoferrins, with antimicrobial activity against oral pathogens. FEMS Microbiol Lett 179:217–222
Bradshaw J (2003) Cationic antimicrobial peptides : issues for potential clinical use. BioDrugs 17:233–240
Riedl S, Zweytick D, Lohner K (2011) Membrane-active host defense peptides–challenges and perspectives for the development of novel anticancer drugs. Chem Phys Lipids 164:766–781
Huang Y, Huang J, Chen Y (2010) Alpha-helical cationic antimicrobial peptides: relationships of structure and function. Protein Cell 1:143–152
Bahar AA, Ren D (2013) Antimicrobial peptides. Pharmaceuticals (Basel) 6:1543–1575
Barzyk W, Rogalska E, Wieclaw-Czapla K (2013) Penetration of milk-derived antimicrobial peptides into phospholipid monolayers as model biomembranes. Biochem Res Int 2013:914540
Boman HG (2003) Antibacterial peptides: basic facts and emerging concepts. J Intern Med 254:197–215
Powers JP, Hancock RE (2003) The relationship between peptide structure and antibacterial activity. Peptides 24:1681–1691
Steiner H, Hultmark D, Engstrom A et al (1981) Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 292:246–248
Bulet P, Stocklin R, Menin L (2004) Anti-microbial peptides: from invertebrates to vertebrates. Immunol Rev 198:169–184
Steinberg DA, Hurst MA, Fujii CA et al (1997) Protegrin-1: a broad-spectrum, rapidly microbicidal peptide with in vivo activity. Antimicrob Agents Chemother 41:1738–1742
Romeo D, Skerlavaj B, Bolognesi M et al (1988) Structure and bactericidal activity of an antibiotic dodecapeptide purified from bovine neutrophils. J Biol Chem 263:9573–9575
Gennaro R, Skerlavaj B, Romeo D (1989) Purification, composition, and activity of two bactenecins, antibacterial peptides of bovine neutrophils. Infect Immun 57:3142–3146
Tossi A, Tarantino C, Romeo D (1997) Design of synthetic antimicrobial peptides based on sequence analogy and amphipathicity. Eur J Biochem 250:549–558
Westerhoff HV, Juretic D, Hendler RW et al (1989) Magainins and the disruption of membrane-linked free-energy transduction. Proc Natl Acad Sci U S A 86:6597–6601
Jiang Z, Vasil AI, Hale JD et al (2008) Effects of net charge and the number of positively charged residues on the biological activity of amphipathic alpha-helical cationic antimicrobial peptides. Biopolymers 90:369–383
Fernandez-Vidal M, Jayasinghe S, Ladokhin AS et al (2007) Folding amphipathic helices into membranes: amphiphilicity trumps hydrophobicity. J Mol Biol 370:459–470
Goumon Y, Strub JM, Moniatte M et al (1996) The C-terminal bisphosphorylated proenkephalin-A-(209-237)-peptide from adrenal medullary chromaffin granules possesses antibacterial activity. Eur J Biochem 235:516–525
Kreil G (1997) D-amino acids in animal peptides. Annu Rev Biochem 66:337–345
Rifflet A, Gavalda S, Tene N et al (2012) Identification and characterization of a novel antimicrobial peptide from the venom of the ant Tetramorium bicarinatum. Peptides 38:363–370
Oman TJ, Boettcher JM, Wang H et al (2011) Sublancin is not a lantibiotic but an S-linked glycopeptide. Nat Chem Biol 7:78–80
Mangoni ME, Aumelas A, Charnet P et al (1996) Change in membrane permeability induced by protegrin 1: implication of disulphide bridges for pore formation. FEBS Lett 383:93–98
Shinnar AE, Butler KL, Park HJ (2003) Cathelicidin family of antimicrobial peptides: proteolytic processing and protease resistance. Bioorg Chem 31:425–436
Zhu S, Gao B (2009) A fossil antibacterial peptide gives clues to structural diversity of cathelicidin-derived host defense peptides. FASEB J 23:13–20
Brogden KA (2005) Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 3:238–250
Sitaram N, Nagaraj R (1999) Interaction of antimicrobial peptides with biological and model membranes: structural and charge requirements for activity. Biochim Biophys Acta 1462:29–54
Belaid A, Aouni M, Khelifa R et al (2002) In vitro antiviral activity of dermaseptins against herpes simplex virus type 1. J Med Virol 66:229–234
van der Weerden NL, Hancock RE, Anderson MA (2010) Permeabilization of fungal hyphae by the plant defensin NaD1 occurs through a cell wall-dependent process. J Biol Chem 285:37513–37520
Moerman L, Bosteels S, Noppe W et al (2002) Antibacterial and antifungal properties of alpha-helical, cationic peptides in the venom of scorpions from southern Africa. Eur J Biochem 269:4799–4810
Park Y, Jang SH, Lee DG et al (2004) Antinematodal effect of antimicrobial peptide, PMAP-23, isolated from porcine myeloid against Caenorhabditis elegans. J Pept Sci 10:304–311
Gordon YJ, Romanowski EG, McDermott AM (2005) A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr Eye Res 30:505–515
Fjell CD, Hiss JA, Hancock RE et al (2012) Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov 11:37–51
Fox JL (2013) Antimicrobial peptides stage a comeback. Nat Biotechnol 31:379–382
Warren TK, Warfield KL, Wells J et al (2010) Advanced antisense therapies for postexposure protection against lethal filovirus infections. Nat Med 16:991–994
Iversen PL, Warren TK, Wells JB et al (2012) Discovery and early development of AVI-7537 and AVI-7288 for the treatment of Ebola virus and Marburg virus infections. Viruses 4:2806–2830
Breaker RR, Li S (2013) Antimicrobial compositions and methods. Google Patents
Zhu WL, Shin SY (2009) Effects of dimerization of the cell-penetrating peptide Tat analog on antimicrobial activity and mechanism of bactericidal action. J Pept Sci 15:345–352
Jung HJ, Jeong KS, Lee DG (2008) Effective antibacterial action of tat (47-58) by increased uptake into bacterial cells in the presence of trypsin. J Microbiol Biotechnol 18:990–996
Vives E, Brodin P, Lebleu B (1997) A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem 272:16010–16017
Elmquist A, Lindgren M, Bartfai T et al (2001) VE-cadherin-derived cell-penetrating peptide, pVEC, with carrier functions. Exp Cell Res 269:237–244
Palm C, Netzereab S, Hallbrink M (2006) Quantitatively determined uptake of cell-penetrating peptides in non-mammalian cells with an evaluation of degradation and antimicrobial effects. Peptides 27:1710–1716
Henriques ST, Costa J, Castanho MA (2005) Translocation of beta-galactosidase mediated by the cell-penetrating peptide pep-1 into lipid vesicles and human HeLa cells is driven by membrane electrostatic potential. Biochemistry 44:10189–10198
Zhu WL, Lan H, Park IS et al (2006) Design and mechanism of action of a novel bacteria-selective antimicrobial peptide from the cell-penetrating peptide Pep-1. Biochem Biophys Res Commun 349:769–774
Pooga M, Kut C, Kihlmark M et al (2001) Cellular translocation of proteins by transportan. FASEB J 15:1451–1453
Nekhotiaeva N, Elmquist A, Rajarao GK et al (2004) Cell entry and antimicrobial properties of eukaryotic cell-penetrating peptides. FASEB J 18:394–396
Scheller A, Oehlke J, Wiesner B et al (1999) Structural requirements for cellular uptake of alpha-helical amphipathic peptides. J Pept Sci 5:185–194
Deshayes S, Gerbal-Chaloin S, Morris MC et al (2004) On the mechanism of non-endosomial peptide-mediated cellular delivery of nucleic acids. Biochim Biophys Acta 1667:141–147
Jung HJ, Park Y, Hahm KS et al (2006) Biological activity of Tat (47-58) peptide on human pathogenic fungi. Biochem Biophys Res Commun 345:222–228
Zhu WL, Hahm KS, Shin SY (2009) Cell selectivity and mechanism of action of short antimicrobial peptides designed from the cell-penetrating peptide Pep-1. J Pept Sci 15:569–575
Viru L, Heller G, Lehto T et al (2011) Novel viral vectors utilizing intron splice-switching to activate genome rescue, expression and replication in targeted cells. Virol J 8:243
Zhuang X, Stahl SJ, Watts NR et al (2014) A cell-penetrating antibody fragment against HIV-1 Rev has high antiviral activity: characterization of the paratope. J Biol Chem 289:20222–20233
Keogan S, Passic S, Krebs FC (2012) Infection by CXCR4-Tropic Human Immunodeficiency Virus Type 1 Is Inhibited by the Cationic Cell-Penetrating Peptide Derived from HIV-1 Tat. Int J Pept 2012:349427
Zhang H, Zhao Q, Bhattacharya S et al (2008) A cell-penetrating helical peptide as a potential HIV-1 inhibitor. J Mol Biol 378:565–580
Zhang H, Curreli F, Waheed AA et al (2013) Dual-acting stapled peptides target both HIV-1 entry and assembly. Retrovirology 10:136
Zhang H, Curreli F, Zhang X et al (2011) Antiviral activity of alpha-helical stapled peptides designed from the HIV-1 capsid dimerization domain. Retrovirology 8:28
Jang S, Hyun S, Kim S et al (2014) Cell-penetrating, dimeric alpha-helical peptides: nanomolar inhibitors of HIV-1 transcription. Angew Chem Int Ed Engl 53:10086–10089
Robaczewska M, Narayan R, Seigneres B et al (2005) Sequence-specific inhibition of duck hepatitis B virus reverse transcription by peptide nucleic acids (PNA). J Hepatol 42:180–187
Abdul F, Ndeboko B, Buronfosse T et al (2012) Potent inhibition of late stages of hepadnavirus replication by a modified cell penetrating peptide. PLoS One 7:e48721
Tiwari V, Liu J, Valyi-Nagy T et al (2011) Anti-heparan sulfate peptides that block herpes simplex virus infection in vivo. J Biol Chem 286:25406–25415
Park PJ, Antoine TE, Farooq AV et al (2013) An investigative peptide-acyclovir combination to control herpes simplex virus type 1 ocular infection. Invest Ophthalmol Vis Sci 54:6373–6381
Otvos L Jr (2000) Antibacterial peptides isolated from insects. J Pept Sci 6:497–511
Otvos L Jr, Cudic M, Chua BY et al (2004) An insect antibacterial peptide-based drug delivery system. Mol Pharm 1:220–232
Gonzalez-Chavez SA, Arevalo-Gallegos S, Rascon-Cruz Q (2009) Lactoferrin: structure, function and applications. Int J Antimicrob Agents 33(301):e301–e308
Gifford JL, Hunter HN, Vogel HJ (2005) Lactoferricin: a lactoferrin-derived peptide with antimicrobial, antiviral, antitumor and immunological properties. Cell Mol Life Sci 62:2588–2598
Duchardt F, Fotin-Mleczek M, Schwarz H et al (2007) A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 8:848–866
Duchardt F, Ruttekolk IR, Verdurmen WP et al (2009) A cell-penetrating peptide derived from human lactoferrin with conformation-dependent uptake efficiency. J Biol Chem 284:36099–36108
Tani A, Lee S, Oishi O et al (1995) Interaction of the fragments characteristic of bactenecin 7 with phospholipid bilayers and their antimicrobial activity. J Biochem 117:560–565
Frank RW, Gennaro R, Schneider K et al (1990) Amino acid sequences of two proline-rich bactenecins. Antimicrobial peptides of bovine neutrophils. J Biol Chem 265:18871–18874
Rousselle C, Smirnova M, Clair P et al (2001) Enhanced delivery of doxorubicin into the brain via a peptide-vector-mediated strategy: saturation kinetics and specificity. J Pharmacol Exp Ther 296:124–131
Chen C, Brock R, Luh F et al (1995) The solution structure of the active domain of CAP18–a lipopolysaccharide binding protein from rabbit leukocytes. FEBS Lett 370:46–52
Larrick JW, Hirata M, Shimomoura Y et al (1993) Antimicrobial activity of rabbit CAP18-derived peptides. Antimicrob Agents Chemother 37:2534–2539
Kaushik N, Basu A, Palumbo P et al (2002) Anti-TAR polyamide nucleotide analog conjugated with a membrane-permeating peptide inhibits human immunodeficiency virus type 1 production. J Virol 76:3881–3891
Ahn DG, Shim SB, Moon JE et al (2011) Interference of hepatitis C virus replication in cell culture by antisense peptide nucleic acids targeting the X-RNA. J Viral Hepat 18:e298–e306
Ganguly S, Chaubey B, Tripathi S et al (2008) Pharmacokinetic analysis of polyamide nucleic-acid-cell penetrating peptide conjugates targeted against HIV-1 transactivation response element. Oligonucleotides 18:277–286
Turner JJ, Ivanova GD, Verbeure B et al (2005) Cell-penetrating peptide conjugates of peptide nucleic acids (PNA) as inhibitors of HIV-1 Tat-dependent trans-activation in cells. Nucleic Acids Res 33:6837–6849
Chaubey B, Tripathi S, Ganguly S et al (2005) A PNA-transportan conjugate targeted to the TAR region of the HIV-1 genome exhibits both antiviral and virucidal properties. Virology 331:418–428
Ahn DG, Lee W, Choi JK et al (2011) Interference of ribosomal frameshifting by antisense peptide nucleic acids suppresses SARS coronavirus replication. Antiviral Res 91:1–10
Yoo JS, Kim CM, Kim JH et al (2009) Inhibition of Japanese encephalitis virus replication by peptide nucleic acids targeting cis-acting elements on the plus- and minus-strands of viral RNA. Antiviral Res 82:122–133
Phalaphol A, Thueng-In K, Thanongsaksrikul J et al (2013) Humanized-VH/VHH that inhibit HCV replication by interfering with the virus helicase activity. J Virol Methods 194:289–299
Xun Y, Pan Q, Tang Z et al (2012) Intracellular-delivery of a single-chain antibody against hepatitis B core protein via cell-penetrating peptide inhibits hepatitis B virus replication in vitro. Int J Mol Med 31:369–376
Mino T, Mori T, Aoyama Y et al (2008) Cell-permeable artificial zinc-finger proteins as potent antiviral drugs for human papillomaviruses. Arch Virol 153:1291–1298
Zhang XM, He DN, Zhou B et al (2013) In vitro inhibition of vesicular stomatitis virus replication by purified porcine Mx1 protein fused to HIV-1 Tat protein transduction domain (PTD). Antiviral Res 99:149–157
Roy A, Chakraborty P, Polley S et al (2013) A peptide targeted against phosphoprotein and leader RNA interaction inhibits growth of Chandipura virus – an emerging rhabdovirus. Antiviral Res 100:346–355
Zhao B, Wang Y, Zhang Y et al (2012) TAT-mediated gp96 transduction to APCs enhances gp96-induced antiviral and antitumor T cell responses. Vaccine 31:545–552
Kugler J, Schmelz S, Gentzsch J et al (2012) High affinity peptide inhibitors of the hepatitis C virus NS3-4A protease refractory to common resistant mutants. J Biol Chem 287:39224–39232
Bocanegra R, Nevot M, Domenech R et al (2011) Rationally designed interfacial peptides are efficient in vitro inhibitors of HIV-1 capsid assembly with antiviral activity. PLoS One 6:e23877
Pan XB, Wei L, Han JC et al (2010) Artificial recombinant cell-penetrating peptides interfere with envelopment of hepatitis B virus nucleocapsid and viral production. Antiviral Res 89:109–114
Meng S, Wei B, Xu R et al (2009) TAT peptides mediated small interfering RNA delivery to Huh-7 cells and efficiently inhibited hepatitis C virus RNA replication. Intervirology 52:135–140
Abes S, Moulton HM, Clair P et al (2006) Vectorization of morpholino oligomers by the (R-Ahx-R)4 peptide allows efficient splicing correction in the absence of endosomolytic agents. J Control Release 116:304–313
Moulton HM, Fletcher S, Neuman BW et al (2007) Cell-penetrating peptide-morpholino conjugates alter pre-mRNA splicing of DMD (Duchenne muscular dystrophy) and inhibit murine coronavirus replication in vivo. Biochem Soc Trans 35:826–828
Yuan J, Stein DA, Lim T et al (2006) Inhibition of coxsackievirus B3 in cell cultures and in mice by peptide-conjugated morpholino oligomers targeting the internal ribosome entry site. J Virol 80:11510–11519
Pärn K, Viru L, Lehto T et al (2013) Transfection of infectious RNA and DNA/RNA layered vectors of semliki forest virus by the cell-penetrating peptide based reagent PepFect6. PLoS One 8:e69659
Gehl J (2003) Electroporation: theory and methods, perspectives for drug delivery, gene therapy and research. Acta Physiol Scand 177:437–447
Dalby B, Cates S, Harris A et al (2004) Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications. Methods 33:95–103
Kato T, Date T, Miyamoto M et al (2003) Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817
Yeh HY, Yates MV, Mulchandani A et al (2008) Visualizing the dynamics of viral replication in living cells via Tat peptide delivery of nuclease-resistant molecular beacons. Proc Natl Acad Sci U S A 105:17522–17525
Caignard G, Bourai M, Jacob Y et al (2009) Inhibition of IFN-alpha/beta signaling by two discrete peptides within measles virus V protein that specifically bind STAT1 and STAT2. Virology 383:112–120
Acknowledgements
This work was supported by the EU through the European Regional Development Fund through the project Tumor-Tech (3.2.1001.11-0008) and Centre of Excellence of Chemical Biology, and by the Estonian Ministry of Education and Research through IUT20–26.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this protocol
Cite this protocol
Pärn, K., Eriste, E., Langel, Ü. (2015). The Antimicrobial and Antiviral Applications of Cell-Penetrating Peptides. In: Langel, Ü. (eds) Cell-Penetrating Peptides. Methods in Molecular Biology, vol 1324. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-2806-4_15
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2806-4_15
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-2805-7
Online ISBN: 978-1-4939-2806-4
eBook Packages: Springer Protocols